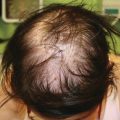
Fig. 3.1
Pseudolymphomatous folliculitis. Indurated erythematous nodule with associated follicular accentuation and scale on the forehead of an adolescent
A special form of CLH that is more common in children and adolescents warranting special mention is acral pseudolymphomatous angiokeratoma of children (APACHE) . This clinical entity is characterized by asymptomatic red-to-violaceous grouped papules with variable overlying keratotic scale, typically at acral locations [12, 13]. Initially thought to be a vascular neoplasm, it is now accepted to represent a form of pseudolymphoma. Despite its name, APACHE has also been reported in adults at non-acral sites [14]. Histologically, APACHE is characterized by a polymorphous dermal infiltrate of lymphocytes, histiocytes, and plasma cells admixed about thick-walled blood vessels with plump endothelial cells. Epidermal changes are variable and may include hyperkeratosis, spongiosis, lymphocytic exocystosis, and vacuolization of the basal layer [12, 13]. Treatment options include excision, intralesional corticosteroid injection, cryotherapy, or radiation therapy [13].
A diagnosis of CLH can only be made via thoughtful synthesis of clinical and histopathologic findings and in some cases may only be diagnosed after prolonged monitoring with regular follow-up. A diagnosis of CLH/pseudolymphoma is suggested by the localized nature of the lesions, an absence of constitutional symptoms, and recognition of an inciting agent. Histopathology with a mixed infiltrate and the absence of marked atypia or a clonal population is also reassuring of a benign process.
Treatment mandates cessation of the inciting agent when CLH is recognized as medication induced [7]. CLH often also responds to biopsy by self-resolving. Beyond observation, however, more active interventions may include topical or intralesional corticosteroids or administration of anti-inflammatory agents such as doxycycline or methotrexate [2]. Treatment with appropriate antibiotics also causes resolution of CLH induced by Borrelia species [8]. In select cases, radiation therapy or surgical excision may also be considered, though this must be carefully weighed against the risks of these procedures, particularly in children. While in general CLH is believed to follow a benign course, it has been proposed that persistent antigenic stimulation may promote evolution to malignancy [3]. As such, regular clinical monitoring via physical and detailed history is warranted for all patients with CLH, even after resolution.
Pityriasis Lichenoides
Key Points
Pityriasis lichenoides (PL) is a benign disorder that exists on a spectrum with both acute and chronic forms.
It is typically self-limited and is not thought to predispose to malignancy but can wax and wane over months to years.
A severe variant of PL known as febrile ulceronecrotic Mucha-Habermann disease is characterized by rapid onset of characteristic lesions with high fevers and systemic involvement; in rare cases it can be fatal.
Pityriasis lichenoides (PL) is a benign lymphoproliferative disorder that is typically classified as two main variants: pityriasis lichenoides et varioliformis acuta (PLEVA) and pityriasis lichenoides chronica (PLC) . These two manifestations lay on a disease spectrum, with PLEVA being more acute and symptomatic and PLC being more chronic in nature, though some patients may exhibit features of both [15, 16]. Both presentations are rare and have a collective incidence of about 1/2000 people per year [17].
The average age of onset of PL is 6.5 years with rare cases reported in infancy, and even at birth, with a slight male predominance [18]. In a review by Hapa et al. of 24 patients (ages 2–14 years, with a median age of 7 years) with PL, PLC was more common than PLEVA (62.5–25%, respectively), with features of both in a small subset of patients (2.5%) [19]. In contrast, a larger retrospective review of 124 patients in 2007 identified PLEVA in 57.3% and PLC in 37% of patients, with features of both seen in the remainder [16]. Patients with PLEVA had a younger median age of onset (60 months) as compared to those with PLC (72 months), with a median duration of disease ranging from 18 months in PLEVA to 20 months in patients with PLC [16]. Both subtypes present more commonly in the spring or the fall [16, 19].
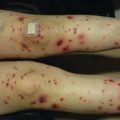
Clinically, PLEVA is characterized by the abrupt onset of pruritic and at times painful, symptomatic papulovesicles with necrotic, ulcerative, or hemorrhagic changes (Fig. 3.2). Early in its course, the condition may be confused with varicella. Patients with PLEVA, however, lack the typical fever and mucous membrane involvement seen in varicella, and the course of PLEVA is considerably more prolonged. In contrast, the hallmark of PLC is recurrent crops of asymptomatic, scaly, erythematous, or red-brown papules and plaques with central adherent scale that flatten or regress over a period of weeks. Lesions tend to be diffuse, though some patients may have truncal predominance, as well as segmental restriction of their disease [20]. Clinically, the papulosquamous appearance of this condition may resemble pityriasis rosea or hypopigmented mycosis fungoides. Rarely, restriction to the palms and soles resembling psoriasis can occur [21]. Lesions of both PLEVA and PLC may be present at all stages of development. In both variants, the lesions usually resolve with hyper- or hypopigmentation [16]. As compared to the clinical presentation in adults, children with PL are more likely to have more prominent dyspigmentation and extensive cutaneous involvement, especially of the face [22].
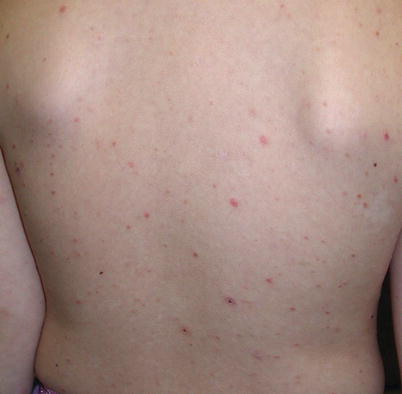
Fig. 3.2
Pityriasis lichenoides et varioliformis acuta. Multiple erythematous papules and vesicles, some with associated ulceration and hemorrhagic crusting, present on the back of a child
Notably, the histopathology of both entities can overlap, with more subtle changes identified in PLC. Tissue examination reveals focal parakeratosis, mild to moderate acanthosis, focal areas of spongiosis, few dyskeratotic keratinocytes, vacuolar interface changes, a mild superficial perivascular lymphocytic infiltrate, and variable erythrocyte extravasation. Necrotic keratinocytes, vesiculation, and ulceration may be seen in the epidermis. Immunohistochemistry also helps to distinguish these entities, as a predominantly CD8+ T-cell lymphocytic infiltrate is present in patients with PLEVA, while the infiltrate in patients with PLC is characterized primarily by CD4+ T-cells [23].
PLEVA and PLC are regarded as benign, reactive disorders, and several theories regarding their etiology have been postulated. Specifically, PLEVA and PLC have been proposed to represent a T-cell-mediated reaction triggered by an infectious agent or medication or to occur secondary to a T-cell dyscrasia. A history of a preceding URI is reported in a third of cases and preceding drug or vaccination in 20% of cases [18, 19]. Suspected infectious triggers include VZV [24], HHV-8 [23], streptococcal pharyngitis, Toxoplasma gondii, parvovirus B19, Epstein-Barr virus (EBV), and human immunodeficiency virus (HIV) [25]. Implicated medications and vaccinations include subcutaneous immunoglobulin [26]; etanercept [27]; measles, mumps, and rubella (MMR) vaccine; influenza vaccine; and hepatitis B immunization [18, 28, 29]. Clonality has been detected in patients with PL, supporting an alternate theory that these entities represent a lymphoproliferative disorder rather than an inflammatory process [30, 31]. Currently, it is unclear if T-cell clonality in PL is dependent upon a specific disease trigger. It is also unknown whether those patients identified as having a clonal population may be at increased risk of developing a secondary lymphoproliferative disease.
PL often waxes and wanes and may exhibit spontaneous resolution. Treatment is initiated due to concerns about the appearance of the eruption, associated symptoms, or to guard against long-term sequelae, such as scarring. First-line treatment options for both PLEVA and PLC include oral antibiotics with or without topical corticosteroids or topical immunomodulators. Erythromycin is commonly administered, with greater than 50% improvement in skin disease in 64% of patients at 1 month, 73% at 2 months, and 83% at 3 months [19]. It is recommended that erythromycin be continued for 2–3 months after resolution given concern for recurrence if stopped too quickly [18]. Cephalexin, amoxicillin-clavulanic acid, cefaclor, tetracycline and minocycline (both contraindicated in children younger than 8 years old), and azithromycin have also been used with some success [32, 33]. Second-line treatment options include ultraviolet B (UVB) and psoralen with ultraviolet A (PUVA) phototherapy , though their success and tolerability may vary based on the age of the patient [34]. PLC may respond better to phototherapy than PLEVA [35]. Narrowband (nb) UVB alone has been successful in 44–48% of patients with PLC [36]. A recent study demonstrated greater efficacy of nbUVB compared to broadband (bb) UVB and PUVA, as recurrence was decreased in patients who received nbUVB therapy. Patients on average achieved a response with 18.8–22 sessions. Commonly observed side effects of phototherapy include erythema, pruritus, and discomfort or pain [37]. PUVA may also be less optimal given its risk of treatment-related secondary cutaneous malignancy [38]. Third-line agents include methotrexate, acitretin, dapsone, or cyclosporine. Up to 77% of patients have evidence of disease recurrence after complete clearance with appropriate treatment [16].
Febrile ulceronecrotic Mucha-Habermann disease (FUMHD) is a rare and severe variant of PLEVA that may follow a diagnosis of PLEVA or occur de novo [39, 40]. When a patient presents with a PLEVA-like eruption with evidence of rapid clinical progression and systemic symptoms, FUMHD is probable [41]. It tends to occur more commonly in children and young adults. In contrast to adults, children may demonstrate a shorter time transforming from PLEVA to FUMHD with more frequent mucosal involvement and a more favorable outcome, [42] though fatal cases have been reported [43]. Affected patients exhibit a rapid onset of necrotic papules coalescing into large ulcerations with histologic features of PLEVA . Mucous membrane involvement is evident in about 28% of cases (including oral, genital, and conjunctival mucosae), with systemic involvement in 45%. Systemic symptoms include markedly high fevers, abdominal pain, diarrhea, arthritis, pulmonary involvement, central nervous system involvement, and sepsis [25, 43, 44]. Laboratory abnormalities may include increased leukocyte count, elevated erythrocyte sedimentation rate and C-reactive protein, anemia, mild hypergammaglobulinemia, hypoproteinemia, hypoalbuminemia, hypocalcemia, eosinophilia, lymphopenia, and positive skin and blood cultures [44]. Clinical mimickers of FUMHD include varicella, papulovesicular pityriasis rosea, leukocytoclastic vasculitis, and lymphomatoid papulosis [44, 45]. Rarely, FUMHD may clinically mimic Stevens-Johnson syndrome (SJS) , given the extent of cutaneous involvement with epidermal necrosis [41]. However, distinguishing features include flexural accentuation, constitutional manifestations, and histologic appearance. Similar to PL, no clear triggers have been identified for FUHMD, though isolated cases have demonstrated seropositivity to VZV and HSV-2 [46, 47]. Treatment modalities for FUMHD may overlap with PL; however, given its severity with rapid progression, systemic complications, and potential fatality, more aggressive or combination medications are utilized. Treatment options include methotrexate [40, 41, 48, 49], TNF-alpha inhibitors [50], prednisone [41], cyclosporine [43], intravenous immune globulin (IVIG), and extracorporeal photopheresis [51].
Lymphomatoid Papulosis
Key Points
Lymphomatoid papulosis (LyP) is a CD30+ cutaneous lymphoproliferative disorder characterized by self-healing but recurrent, ulcerative papules.
Treatment of LyP is not thought to influence disease course or progression.
LyP is associated with secondary malignancies in approximately 10–20% of patients.
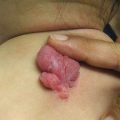
Lymphomatoid papulosis (LyP) is a chronic primary cutaneous CD30+ lymphoproliferative disorder of intermediate malignant potential. Clinically it is distinguished by crops of pink-red papulonodules, most commonly distributed on the trunk and extremities (Fig. 3.3). LyP characteristically advances over weeks to months through stages of necrosis, ulceration, and hemorrhagic crusting before spontaneously resolving with pigmentary changes and scarring. Individual lesions are often in different stages of evolution [52]. A variant of LyP termed persistent agminated LyP (PALP) presents as multiple lesions of LyP within a circumscribed area that wax and wane in intensity but never completely resolve. As a result of this behavior, whether PALP represents a localized form of LyP or a distinct lymphoproliferative disorder on the spectrum of mycosis fungoides (MF) remains controversial [53, 54].
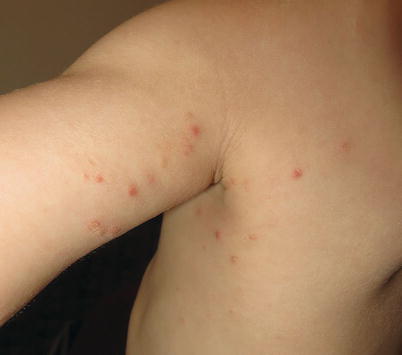
Fig. 3.3
Lymphomatoid papulosis. A crop of pink-red papulonodules with early scale and crust present on the arm and axilla of a child
Although more common in older adults, LyP is also well documented in children, and its incidence may be underestimated [55]. Recent retrospective analyses and systematic literature reviews provide greater insight into the nature of pediatric LyP. In children, LyP occurs at a mean age of approximately 7–8 years, though diagnosis is often delayed by 1 year [55, 56]. Boys are somewhat more frequently afflicted than girls [55, 56]. As in adults, pediatric LyP favors the extremities and trunk, though generalized forms are not uncommon and multiple lesions (up to 75 total in some reported cases) may be seen [57]. In a series of 25 children with LyP, the majority of patients had fewer than 10 lesions that resolved over a mean time of 5 weeks, though disease duration was, on average, 18 months [55]. Approximately 25% of all patients had recurrent disease, with disease-free intervals between outbreaks lasting from less than 1–20 months [55]. Interestingly, though LyP is typically thought to be asymptomatic, children often endorse associated pruritus that may lead to a misdiagnosis of arthropod assault [55, 56].
LyP can be classified histologically into different subtypes, denoted LyP types A through F [52]. Although the nuances of each histologic subtype are beyond the scope of this text, some salient features are worth noting. Among both children and adults, type A LyP appears to be the most common form [55–58]. Type A LyP demonstrates a wedge-shaped infiltrate of grouped, large, pleomorphic lymphocytes admixed among neutrophils, eosinophils, and histiocytes. These tumor cells usually possess a CD4+ T-helper cell phenotype with CD30 expression, though many cases of CD8+ LyP have been reported in the pediatric population, without evident prognostic implications [57, 58]. Type B LyP displays a superficial perivascular or band-like infiltrate of lymphocytes with cerebriform nuclei in the dermis accompanied by lymphocytic epidermotropism that is reminiscent of the histologic features of MF (see below). In this subtype, many neoplastic cells lack CD30 expression, and distinction from MF on histologic grounds alone can be difficult, requiring clinicopathologic correlation. Type C LyP is characterized by sheets of large, atypical, CD30+ lymphocytes and resembles anaplastic large-cell lymphoma [58]. Distinguishing between these two entities is difficult and requires clinicopathologic correlation. Rare, histologically unique subtypes D, E, and F have also been reported [59]. T-cell clonality is common in lesions of LyP [55, 58, 60]. Studies conflict as to whether the histologic subtype of LyP influences clinical course or prognosis, though a retrospective study of 123 patients with LyP suggests that T-cell clonality and multiple subtypes of LyP present in the same individual may indicate an increased risk of associated hematologic malignancy [61].
The etiology of LyP is unknown, though persistent antigenic stimulation of skin-resident T cells has been suggested. In support of this, up to 25% of pediatric patients who develop LyP report having an antecedent viral illness prior to disease onset, while another quarter of patients carry a diagnosis of atopic dermatitis [55, 56]. PL can also be seen concomitantly with LyP. Given that some cases of PL are identified as having a CD30+ cellular infiltrate, it has been proposed that PL and LyP are related entities, though this remains controversial [55, 56]. Importantly, approximately 10–20% of patients with LyP develop a secondary hematologic malignancy prior to, concurrently with, or after their diagnosis. MF, anaplastic large cell lymphoma (ALCL) , or Hodgkin disease are the most commonly associated malignancies [52, 62]. Although MF is the most frequent LyP-associated malignancy in adults, ALCL appears to be the most common secondary hematologic malignancy in children [52, 56, 62, 63].
Initial evaluation of LyP includes skin biopsy, complete blood count (CBC) with differential, a comprehensive metabolic panel (CMP) , and lactate dehydrogenase (LDH) . If malignancy is considered in the differential diagnosis, more extensive evaluation may be warranted. LyP follows a spontaneous, self-resolving course. Thus, providers must carefully weigh risks and benefits when considering treatment. Treatment does not appear to influence the natural course of the condition or development of associated malignancies; no known “curative” therapies exist [52, 56, 64]. Therefore, close observation is a reasonable option in the management of LyP. Factors that may warrant active intervention include multiple cosmetically bothersome lesions, intense pruritus, or a desire to reduce the risk of subsequent scarring. For localized, infrequently recurrent disease, application of ultrapotent topical corticosteroids can help speed resolution or limit growth and ulceration of individual lesions [52]. For more widespread disease, systemic agents are more appropriate options, but their specific risks must be carefully considered in the pediatric population. For example, PUVA appears to be a useful treatment for LyP. However, given its risk for inducing cutaneous malignancy, nbUVB therapy may be a more appropriate choice in children, despite only anecdotal evidence to support its efficacy [52, 62]. Other potential treatment options include oral antibiotics (i.e., doxycycline), though low-dose methotrexate may be a safe, more effective, and therefore preferred option [64]. Topical nitrogen mustard, as well as systemic bexarotene or interferon, may also be considered as second-line agents [52]. Owing to the risk of developing a secondary malignancy, all patients with LyP warrant regular, long-term monitoring.
Cutaneous T-Cell Lymphoma/Mycosis Fungoides
Key Points
Mycosis fungoides (MF) is the most common subtype of cutaneous T-cell lymphoma (CTCL) in children.
The hypopigmented variant is the most common form of pediatric MF.
Most children with MF have both limited body surface area with rare systemic involvement and have life expectancies similar to age-matched controls.
Cutaneous T-cell lymphoma (CTCL) is a general term that refers to primary extranodal T-cell neoplasms of the skin. The frequency of CTCL is estimated at 10.2 cases per million, which appears to have stabilized after many years of increasing incidence [65]. Although much more common in adults, the entire spectrum of CTCL has been reported in children [66].
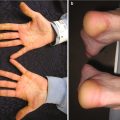
Among the assorted forms of CTCL, MF is the most common variant in both adults and children. MF can present in diverse ways, from limited patch-stage disease to frank erythroderma, with variable extracutaneous involvement. Classically, MF presents as scaly, erythematous patches or plaques that favor sun-protected, “double-covered” areas including the buttocks and upper thighs (Fig. 3.4). Overlying skin atrophy may be associated. More advanced stages, which are uncommon in children, are characterized by larger tumors with a propensity toward ulceration or erythroderma with accompanying constitutional symptoms. Staging guidelines for MF are found in Table 3.1.
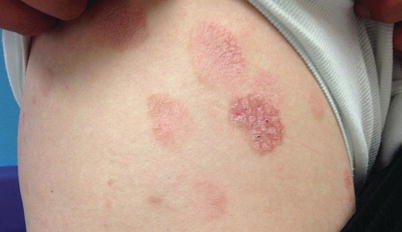
Fig. 3.4
Mycosis fungoides. Nummular, scaly, erythematous patches and thin plaques present on the upper thigh and buttocks of a teenager
Skin | Description | |||
|---|---|---|---|---|
TNMB classification | ||||
T1 | Patches, papules, and/or plaques covering <10% total body surface area | |||
T2 | Patches (T2a), papules, and/or plaques (T2b) covering ≥10% total body surface area | |||
T3 | One or more tumors (≥1 cm in diameter) | |||
T4 | Erythroderma (erythema involving ≥80% total body surface area) | |||
Nodes | ||||
N0 | No clinically abnormal lymph nodes identified | |||
N1 | Atypical lymph nodes, histopathology Dutch grade 1 or NCI LN0–2 | |||
N2 | Atypical lymph nodes, histopathology Dutch grade 2 or NCI LN 3 | |||
N3 | Atypical lymph nodes, histopathology Dutch grades 3–4 or NCI LN 4 | |||
NX | Abnormal lymph nodes without histopathologic evaluation | |||
Visceral | ||||
M0 | No visceral organ involvement | |||
M1 | Visceral involvement with histologic confirmation (specify organ) | |||
MX | Abnormal viscera without histologic confirmation | |||
Blood | ||||
B0 | Absence of significant blood involvement; ≤5% of peripheral blood lymphocytes are Sezary cells, <15% CD4+/CD26 or CD4+/CD7 cells of total lymphocytes | |||
B1 | Low blood tumor involvement; >5% of peripheral blood lymphocytes are Sezary cells or ≥15% CD4+/CD26 or CD4+/CD7 cells of total lymphocytes but do not meet classification as B0 or B2 | |||
B2 | High blood tumor involvement; ≥1000/mcL Sezary cells or CD4/CD8 ≥ 10 or ≥40% CD4+/CD7 or ≥30% CD4+/CD26 cells of total lymphocytes | |||
Clinical staging of mycosis fungoides | ||||
T | N | M | B | |
IA | 1 | 0 | 0 | 0–1 |
IB | 2 | 0 | 0 | 0–1 |
IIA | 1–2 | 1–2 | 0 | 0–1 |
IIB | 3 | 0–2 | 0 | 0–1 |
IIIA | 4 | 0–2 | 0 | 0 |
IIIB | 4 | 0–2 | 0 | 1 |
IVA1 | 1–4 | 0–2 | 0 | 2 |
IVA2 | 1–4 | 3 | 0 | 0–2 |
IVB | 1–4 | 0–3 | 1 | 0–2 |
Other presentations of MF that have been described in children include pigmented purpuric, pityriasis lichenoides-like, isolated acral (Woringer-Kolopp), and leukemic variants [67–70]. Among children, the hypopigmented variant of MF is the most common presentation and is overrepresented relative to its incidence in adults [67, 68, 71–73]. This form is characterized by hypopigmented macules and patches, usually without secondary change, and can occur in isolation or alongside more traditional papulosquamous lesions of MF. Although more commonly recognized in darker-skinned children, hypopigmented MF is known to occur in Caucasian children, as well [68, 74, 75]. Irrespective of clinical appearance, most children present with early stage disease (IA, IB, or IIA) [67, 68, 71, 74].
Folliculotropic MF (FMF) is a rare variant that is more common in adults and clinically is characterized by acneiform lesions including comedones, milia, or cysts. Hairless patches and plaques with folliculocentric papules and variable degrees of induration are a more common presentation in children [76]. FMF deserves special consideration because its clinical appearance often overlaps with that of benign, reactive follicular mucinosis (FM). This distinction is vital as FMF in adults appears to carry a worse prognosis than classic MF, and both treatment and prognosis differ considerably between FM and FMF [77]. Historically, the clinical presentation of FM was divided into two main groups: FM limited to the head and neck in young patients and more widespread FM that was more typically seen in older adults. Importantly, the former usually follows an indolent course, while more widespread disease is thought to be more aggressive, with a greater likelihood of transformation into FMF [78, 79]. T-cell clonality does not appear to help distinguish between FM and FMF and may be of minimal prognostic value [80].
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


