Fig. 9.1
Molecular organisation of laminin 332 in the dermal–epidermal junction. (a) Structure of human laminin 332 (Modified from Rousselle and Beck [85]). Laminin 332 is composed of three subunits: α3A, β3 and γ2. The large LG structure located at the C-terminal end of the α3 chain contains five repeating LG domains. The first three repeats (LG1–LG3) interact with α3β1, α6β1 and α6β4 integrins, while the last two (LG45) contain binding sites for syndecan-1 and syndecan-4. Laminin 332 is synthesised as a precursor molecule that undergoes maturation by proteolytic processing at the α3Α chain N- and C-terminus as well as at the γ2 chain N-terminal extremity. The cleavage sites are indicated by arrows as well as enzymes involved identified so far. (b) Model for laminin 332 assembly at the dermal–epidermal junction. Theoretical assembly of laminin-332 and laminin-311 in the anchoring complexes
The second mechanism reports a direct interaction between anchoring filaments and anchoring fibrils. Anchoring fibrils are disulphide bond–stabilised dimers of type VII collagen [13]. Monomeric laminin 332 as well as the laminin 332/laminin 311 dimer directly bind the amino-terminal globular domain NC1 of type VII collagen, and the interaction is likely to occur within the short arm of the β3 and/or γ2 subunit [21, 87] (Fig. 9.2). Maturation is therefore important for the function of laminin 332 in the establishment and maintenance of the skin basement membrane structural integrity. However, recent studies have been conducted to elucidate the role of the cleavable domains in precursor laminin 332 [85]. A potential function for the tandem LG45 domains was suspected based on the ability of laminin 332 to trigger distinct cellular events depending on the level of processing of its α3 chain. While laminin 332 that lacks LG45 is found in mature basement membranes where it has an important function in the nucleation and maintenance of HDs through α3β1 and α6β4 integrin interactions [37, 52, 108], laminin 332 with intact LG45 (α3200) was found in migratory/remodelling situations such as epidermal repair [32, 89]. Epidermal injury activates the transcription and deposition of laminin 332 into the provisional matrix by the leading keratinocytes in the process of epidermal outgrowth and migration at the wound edge [44, 52, 90]. Noteworthy, α3200 laminin 332 is found in this provisional matrix but is absent from mature basement membranes [37, 52, 98]. Laminin 332 with an α3200 chain is also found in the ECM of migrating keratinocytes in vitro [26, 77]. Recently, laminin 332 comprising an α3200 chain was proposed to be involved in the invasion of squamous cell carcinomas in vivo [105].
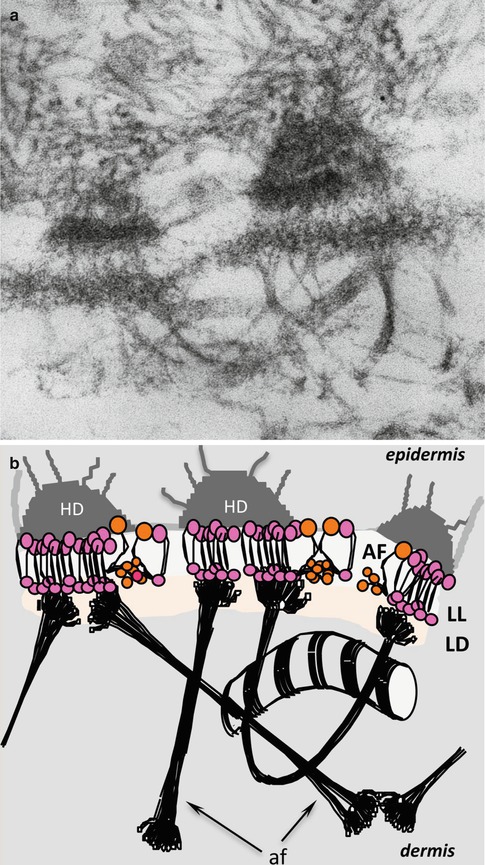
Fig. 9.2
Supramolecular organisation of anchoring complexes. (a) Transmission electron microscopy micrograph of human skin allowing the visualisation of the lamina lucida (LL), lamina densa (LD), hemidesmosome (HD), anchoring filaments (AF) and anchoring fibrils (af), bar 100 nm. (b) Schematic representation of anchoring complexes. Laminin 332 (pink) and laminin 311 (orange) within the anchoring filaments (AF) connect to hemidesmosomes (HD), span the lamina lucida and interact with anchoring fibrils (af) within the lamina densa (LD). Dermal anchoring fibrils are made of type VII collagen. This representation focuses only on anchoring complexes and does not show the molecular network formed with collagen IV, nidogen, laminin 511 and perlecan, which also significantly contributes to the structural properties of the dermal–epidermal junction
LG45 in precursor laminin 332 was suggested to have a function in the deposition of laminin 332 [98, 105]. A study reported a missense mutation in the LAMA3 gene affecting LG4 in a patient with a mild non-Herlitz JEB phenotype [94]. This mutation resulting in G1506E triggers an imperfect local protein folding which, without impairing trimerisation of the coiled coil, causes laminin 332 intracellular accumulation within the endoplasmic reticulum. Only a small amount of the laminin 332 harbouring the mutated α3 chain is secreted and physiologically processed, thus providing partial adhesion functions and explaining the mild phenotype. Therefore structural changes caused by mutations of this highly conserved residue throughout laminin LG4 domains highlight potential important functions of LG45 in laminin secretion. It was also proposed that precursor laminin 332, together with integrin α3β1, plays a central role in cell migration [32, 38, 79]. Syndecans have been described as cellular receptors potentially involved in these mechanisms [5, 16, 79, 101, 109]. Information concerning the precursor γ2155 chain is low; however its involvement in keratinocyte migration was suggested [26, 75, 76].
9.3 Junctional Epidermolysis Bullosa
The important role of laminin 332 and of the α6β4 integrin in epidermal–dermal adhesion was early confirmed by the finding that mutations affecting either of these molecules cause JEB, a genetic disorder that affects skin and mucous membranes [17]. It is characterised by a mesenchymal–epithelial separation within the lamina lucida and by hemidesmosomal abnormalities. JEB is generally divided into three subtypes: Herlitz (lethal), non-Herlitz (nonlethal) and JEB with pyloric atresia [4, 29, 35, 114]. All of them are inherited in an autosomal recessive manner [93] except for a single case recently reported [2]. Herlitz JEB is caused by a complete absence of laminin 332 [6, 65, 81], while non-Herlitz is caused by missense mutations leading to a reduction in functional laminin 332 or complete absence of collagen XVII [6, 65, 81, 115]. JEB with pyloric atresia is caused by a genetic mutation of α6 or β4 subunits that are the main receptors for laminin 332 located beneath HDs [91, 114]. The majority of mutations identified to date in both H-JEB and nH-JEB reside in the three genes that encode the αβγ chains of laminin 332, LAMA3α, LAMB3 and LAMC2, respectively [74, 111]. Normally, these three genes are expressed during all developmental stages of the skin including all steps of adhesion, proliferation and differentiation [31]. Nonsense or frameshift mutations of any of the three laminin 332 genes causing a premature termination codon (PTC) usually lead to a decrease in transcript levels due to nonsense-mediated mRNA decay [40]. If such mRNAs are translated, truncated polypeptides are produced that are incapable of participating in the formation of a functional heterotrimer. Other mutations that may occur in the laminin 332 genes include missense mutations, exon-skipping mutations and in-frame insertions or deletions. In these cases, translation of the resulting mRNA results in structurally imperfect protein production, which still allows secretion of more or less functional laminin 332 [96]. Most mutations related to the JEB are found in the LAMB3 gene and mainly in the exons encoding the N-terminal part of the β3 chain. The LAMB3 portion coding for the C-terminal α-helical domain, which is involved in subunit assembly, contains much less mutations [73, 97].
There are particularly two recurrent “hotspot” LAMB3 mutations, R635X and R42X, leading to PTCs which account for half of all LAMB3 mutations and result from a C-to-T transition [49, 73, 82]. In the LAMC2 gene, most of the mutations are found within the LE domain and the L4 module and rarely in the C-terminal α-helical domain [96]. Some mutations are also found within the LG1–LG5 globular domains of the LAMA3 gene. Particularly, the LG3 mutation K1299X, which affects the charged amino acid lysine at position 1299, is related to a severe phenotype seen in the homozygous human patients [45, 73, 78]. As the α3 chain is found in laminins 311 and 321, mutations in LAMA3 gene also affect these two laminins’ functions [96]. Based on the clinical severity, JEB patients with laminin 332 mutations have been classified into the Herlitz and non-Herlitz types, and there are some general “rules” regarding phenotype–genotype correlations [30]. In general terms, patients harbouring PTC causing mutations on both alleles (homozygous or compound heterozygous) suffer from the severe H-JEB [1, 81]. As a result of these mutations, the synthesis of one laminin 332 chain is abolished and no functional trimeric laminin 332 is produced [66, 80, 83]. The PTC mutations in H-JEB result in the absence of laminin 332 within the dermal–epidermal junction as confirmed by staining with laminin 332–specific monoclonal antibodies [39, 68, 113]. Examination of skin biopsies from H-JEB patients by electron microscopy reveals small rudimentary or absent HDs, as well as cleavages through the lamina lucida [103]. The fact that laminin 332 is involved in the regulation of motility and proliferation of keratinocytes predicts that its absence can affect the normal wound-healing process causing chronic erosions and formation of granulation tissue. All resulting laminin 332 defects cause a generalised blistering and early death of the patient, usually during the first year of life. The epidermis of an affected infant can be extensively peeled away only on simple handling, while in many cases there is death by overwhelming infection, caloric deprivation or from organ failure deriving from complications of the disease [73]. nH-JEB patients often carry a PTC on one allele and a missense or in-frame splice site mutation on the alternate allele. In these cases there is often a milder phenotype due to the production of partial or abnormal laminin 332. This is in accordance with immunofluorescence mapping studies showing diminished expression of laminin 332 within the dermal–epidermal junction of these patients [116]. The nH-JEB phenotype is characterised by lifelong blistering in a distribution that predominates in sites exposed to friction, trauma or heat, atrophic scars, hypopigmentation or hyperpigmentation at sites of healed blisters, incomplete alopecia, dystrophic nails and dental abnormalities [24, 30, 116]. The course and prognosis of nH-JEB depend on how severely affected the target protein is. Thus, some patients have a mild phenotype and normal lifespan (nH-JEB localised), and others have a severe disease and an increased risk of squamous carcinoma development and death (nH-JEB generalised) [46, 47].
9.4 Mucous Membrane Pemphigoid
Mucous membrane pemphigoid (MMP) is a heterogeneous group of rare autoimmune blistering diseases and is characterised by the presence of autoantibodies to various components of the dermal–epidermal anchoring complex causing erosive lesions followed by scarring of the skin and mucous membranes [50]. On the basis of the 2002 international consensus [19], MMP includes blistering diseases with preferential mucous membrane involvement. Patients with MMP may exhibit linear deposits of IgG and/or IgA autoantibodies and/or complement fragments in epithelial basement membranes. Subtypes of MMP correspond to the nature of the antigen targeted such as BP230 and BP180, laminin 332 and laminin 311, type VII collagen or the α6 and β4 integrin subunits [see Caux and Prost, Part III, Chapter 14; 50]. Early studies revealed that laminin 332 is a target antigen in about 25 % of MMP patients and most patients have IgG antibodies against the α-subunit of the heterotrimer [48, 55] that recognise both laminin 332 and laminin 311 [20]. The pathogenicity of anti-laminin 332 antibodies has been documented in vitro and in vivo. Passive transfer of anti-laminin 332 IgG to neonatal or adult mice induced subepidermal blisters of the skin and mucous membranes that mimicked clinical, histological and immunopathologic features seen in MMP patients [58]. Mice injection of Fab fragments directed against laminin 332 produced the same results as well as injection in mice lacking complement, mast cells or T cells, suggesting that the antibodies induced epidermal detachment in a noninflammatory and direct manner [53, 58]. An experimental human skin graft model was also used and revealed that patients’ anti-laminin 332 autoantibodies induced subepidermal blisters [54]. Assaying for anti-laminin 332 reactivity is of particular importance as 25 % of patients with laminin 332-specific antibodies are suspected to develop a malignancy [28, 59]. While direct immunofluorescence microscopy of patients’ epidermis and mucosal epithelium is still the gold standard for the diagnosis of pemphigoid diseases, diagnosis can be made serologically today [57, 95]. Patients with anti-laminin 332 antibodies display circulating IgG autoantibodies that bind to the dermal side of 1 M salt-split skin by indirect immunofluorescence [59]. Western blot analysis has revealed that anti-laminin 332 autoantibodies most often react with the α3 chain [48, 55] and less frequently with the β3 and/or the γ2 chain [33, 34, 55, 64, 102]. However, the exact mechanism is not known yet; it is most likely that antibodies against the α3 chain are disrupting the interaction with the keratinocyte integrins. Moreover, it is possible that autoantibodies against the β3 and γ2 chains may disrupt the interaction between laminin 332 and type VII collagen, as laminin-β3 is thought to mediate this binding [21, 87]. Neither specific epitopes nor laminin domains targeted by anti-laminin 332 autoantibodies have been precisely identified so far, and the use of recombinantly produced laminin domains will probably be helpful in such autoantibodies’ characterisation. Immunoprecipitation with radiolabelled human keratinocytes has been reported to be the most sensitive technique for the detection of serum autoantibodies against laminin 332, and immunoblotting using human keratinocytes’ ECM or purified laminin 332 was used as a convenient method to identify the laminin subunit involved [20, 41, 43, 56]. Enzyme-linked immunosorbent (ELISA)–based assays will probably soon replace these time- and reagent-consuming methods. Some studies conducted so far have reported patients’ serum samples analysis in ELISA assays using either purified laminin 332 or keratinocyte ECM as a substrate [11, 12, 56]. The most recent study [12] reported the analysis of serum samples from 154 patients with MMP and 89 control individuals. The authors found that 20 % of the MMP patients tested had serum anti-laminin 332 autoantibodies. Interestingly, these appeared to be present in a subset of patients with a severe form of the disease.
9.5 Conclusion
Dermatological pathologies related to laminin 332 are most often severe and disabling, highlighting the important structural role of this multifunctional basement membrane protein. Intense research conducted in the last few years has deciphered its molecular characterisation, leading to a better understanding of pathophysiology. Future studies will probably allow discovery of additional unexpected functions for this protein. For instance, a recent study has reported that the expression of laminin 332 at the dermal–epidermal junction enables a precise control of mechanosensitivity of the sensory nerve endings that enter the epidermal layer to contact keratinocytes [23]. The laminin 332–mediated local suppression of axonal branching and mechanotransduction was suggested to prevent hypersensitivity of sensory axons entering the epidermis, therefore playing a role in pain transmission. From its identification in the 1990s [15, 71, 88, 113] to nowadays, laminin 332 has stimulated an increasing interest in the scientific community due to its multifunctional properties and its involvement in human physiological processes and pathologies.
Acknowledgements
Original work by the authors was financially supported by the Agence Nationale grant (ANR-08-PCVI-0031, ANR-13-RPIB-0003-01).
References
1.
Aberdam D, Galliano MF, Vally J, et al. Herlitz junctional epidermolysis bullosa is linked to mutations in the gene (LAMC2) for the gamma 2 subunit of nicein/kalinin (LAMININ-5). Nat Genet. 1994;6:293–7.
2.
Almaani N, Liu K, Dopping-Hepenstal PJC, Lovell PA, Lai-Cheong JE, Graham RM, Mellerio JE, McGrath JA. Autosomal dominant junctional epidermolysis bullosa. Br J Dermatol. 2009;160:1094–7.PubMed
3.
Amano S, Scott IC, Takahara K, Koch M, Champliaud MF, Gerecke DR, et al. Bone morphogenetic protein 1 is an extracellular processing enzyme of the laminin 5 γ2 chain. J Biol Chem. 2000;275:22728–35.PubMed
4.
Anton-Lemprecht I, Schnyder U. Ultrastructure of epidermolysis with junctional blister formation (author’s transl). Dermatologica. 1979;159:377–82.
5.
Araki E, Momota Y, Togo T, Tanioka M, Hozumi K, Nomizu M, et al. Clustering of syndecan-4 and integrin β1 by laminin α 3 chain-derived peptide promotes keratinocyte migration. Mol Biol Cell. 2009;20:3012–24.PubMedCentralPubMed
6.
Ashton G, Mellerio J, Dunnill M, et al. A recurrent laminin 5 mutation in British patients with lethal (Herlitz) junctional epidermolysis bullosa: evidence for a mutational hotspot rather than propagation of an ancestral allele. Br J Dermatol. 1997;136:674–7.PubMed
8.
Aumailley M, Bruckner-Tuderman L, Carter WG, Deutzmann R, Edgar D, Ekblom P, et al. A simplified laminin nomenclature. Matrix Biol. 2005;24:326–32.PubMed
9.
Baudoin C, Fantin L, Meneguzzi G. Proteolytic processing of the laminin α3 G domain mediates assembly of hemidesmosomes but has no role on keratinocyte migration. J Invest Dermatol. 2005;125:883–8.PubMed
10.
Beck K, Dixon TW, Engel J, Parry DA. Ionic interactions in the coiled-coil domain of laminin determine the specificity of chain assembly. J Mol Biol. 1993;231:311–23.PubMed
11.
Bekou V, Thoma-Uszynski S, Wendler O, Uter W, Schwietzke S, Hunziker T, et al. Detection of laminin 5-specific auto-antibodies in mucous membrane and bullous pemphigoid sera by ELISA. J Invest Dermatol. 2005;124:732–40.PubMed
12.
Bernard P, Antonicelli F, Bedane C, Joly P, Le Roux-Villet C, Duvert-Lehembre S, et al. Prevalence and clinical significance of anti-laminin 332 autoantibodies detected by a novel enzyme-linked immunosorbent assay in mucous membrane pemphigoid. JAMA Dermatol. 2013;149:533–40.PubMed
13.
Burgeson RE. Type VII, collagen, anchoring fibrils and epidermolysis bullosa. J Invest Dermatol. 1993;101:252–5.PubMed
14.
Capt A, Spirito F, Guaguere E, Spadafora A, Ortonne JP, Meneguzzi G. Inherited junctional epidermolysis bullosa in the German pointer: establishment of a large animal model. J Invest Dermatol. 2005;124:530–5.PubMed
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


