Fig. 10.1
Distribution and domain structure of kindlin-1. (a) Kindlin-1 (green, arrows) is demonstrated in keratinocytes at focal adhesions and in the epidermis at the dermal-epidermal junction by indirect immunofluorescence staining with specific antibodies. Nuclei are in blue. (b) Schematic representation of the structure of kindlin-1 with FERM subdomains (F0–F3), PH domain, phosphorylation sites (P) and integrin binding site
Expression of kindlin-1 in mice was investigated by RT-PCR, Northern blot and in situ hybridisations during embryonic development and in adult animals. Kindlin-1 was hardly detected in the embryo, with only weak signals in the epithelium of the gut from E12.5 onwards, as well as in the oral epithelium and oesophagus [11]. In adult mice, kindlin-1 transcripts were detected in the bladder and colon and at lower levels in the kidney, skin, small intestine, stomach and thymus [6].
In human adult tissues, kindlin-1 is expressed in the skin, periodontal tissues, intestinal epithelium and kidney [3, 7, 9]. In the skin and oral mucosa, kindlin-1 is localised in basal epidermal keratinocytes in a polarised manner facing the basement membrane (Fig. 10.1a) [9, 12]. In the colon and rectum, kindlin-1 is localised to the plasma membrane of the epithelial cells, co-localised with ezrin and β-catenin [7].
Kindlin-1 expression is influenced by TGF-β levels [13], whereas kindlin-1 phosphorylation is responsive to epidermal growth factor treatment (own unpublished results).
10.2.2 Domain Structure and Interactions
By means of its FERM (four point one ezrin, radixin and moesin) and pleckstrin homology (PH) domains (Fig. 10.1b), kindlin-1 is localised to integrin-linked adhesion sites and provides linkage of filamentous actin in the cell cortex to membrane proteins on the surface of cells [14, 15]. The FERM domain is subdivided in the subdomains F0–F3 and has high homology to the talin FERM domain [13] (Fig. 10.1b). The N-terminal F0 subdomain is required for integrin activation [16, 17], whereas the F3 subdomain interacts directly with β integrin cytoplasmic tails through phosphotyrosine binding folds [16, 18]. Using integrin pull-down assays, kindlin-1 was shown to bind to the membrane-distal NxxY motif and a preceding threonine-containing region in the C-terminus of β integrin cytoplasmic tails, thus modulating integrin activation [11, 19]. The interactions of kindlin-1 with migfilin, integrin-linked kinase, α-actinin and focal adhesion kinase may represent the link between integrins and the actin cytoskeleton [8, 11, 20]. In keratinocytes, kindlin-1 exists in two forms, non-phosphorylated and phosphorylated, and phosphorylation depends on casein kinase 2 [12]. In a global approach to identify proteins whose phosphorylation is cell cycle regulated, two serine (Ser170 and Ser179) phosphorylation sites were identified in kindlin-1 in HeLa cells [21] (Fig. 10.1b).
10.2.3 Kindlins and Integrin Activation
Integrins are members of a large family of functionally conserved receptors which play important roles in cell adhesion, migration, proliferation and survival [22]. Integrins activate intracellular signalling pathways after binding to their ligands (“outside-in” signalling), and they can shift from a low- to a high-affinity state for the ligands (“inside-out” signalling), which is known as “integrin activation” [15]. Talin and kindlins bind to integrin β subunit cytoplasmic tails and cause activation [15]. This is particularly important during development, in response to injury and during inflammation [15]. Deletion of kindlin-2 results in death at implantation, due to defective integrin function, in cells of the endoderm and epiblast [23, 24]. Integrin activation adjusts adhesion and migration of leucocytes and thrombocytes, and its ablation due to kindlin-3 loss-of-function mutations causes leucocyte adhesion deficiency-III with severe bleeding and life-threatening infections [25, 26]. The physiological significance of integrin activation in adult tissues composed of cells with stable adhesion, like the keratinocytes in the epidermis, might be different. Nevertheless, deletion of kindlin-1 significantly reduces integrin activation in intestinal epithelial cells and epidermal keratinocytes [11, 27, 28].
Talin and kindlins accomplish distinct functions in integrin activation [29, 30]. The two NPxY motifs of β1 integrin which serve as binding sites for talin and kindlins seem to play distinct roles in epithelial cells. Binding of talin to the membrane-proximal NPxY is crucial for connecting α5β1 to the actin cytoskeleton and thus permits the tension required for fibronectin fibrillogenesis and cell migration, whereas binding of kindlin to the membrane-distal NPxY regulates α5β1 surface expression and degradation [30]. The kindlin binding site in the β1 integrin cytoplasmic domain serves as a molecular switch enabling the sequential binding of two FERM-domain-containing proteins in different cellular compartments. When β1 integrins are at the plasma membrane, kindlins control ligand-binding affinity. However, when they are internalised, kindlins dissociate from integrins, which are recycled back to the cell surface [18].
10.2.4 Kindlin-1 and Kindlin-2
Whether kindlins accomplish overlapping or distinct functions remains an interesting issue. Keratinocytes express both kindlin-1 and kindlin-2, but nonetheless, in the absence of kindlin-1 in Kindler syndrome, kindlin-2 cannot fully compensate and rescue the defect. Knockdown of either of the kindlins affects adhesion, survival and migration of the keratinocytes and integrin activation, suggesting that kindlins have overlapping functions. Beyond that, sole deficiency of kindlin-2 strongly impairs the formation of cell-cell adhesions [28]. Using integrin β1 knockout keratinocytes as a model, a further difference between kindlin-1 and kindlin-2 has been uncovered; while kindlin-1 and kindlin-2 both bind and co-localise with β1 integrins, only kindlin-1 binds with β6 integrins [10]. Nevertheless, the presence of at least one kindlin partially assures keratinocyte functions, whereas loss of both kindlins has a cumulative impact on cell adhesion, survival and migration and on integrin activation [28].
Another interesting finding regarding the interplay between kindlins was recently reported in neurons. Although in physiological conditions kindlin-2, but not kindlin-1, is present in the nervous system, expression of kindlin-1 enhances integrin activation and signalling and promotes axon regeneration and recovery of sensory functions after dorsal root crush [31].
10.2.5 Kindlin-1 Knockout Mouse Model
The phenotype of the kindlin-1 knockout mice does not recapitulate the human disorder [11]. Animals appear normal at birth, but 2 days postnatal they become dehydrated, fail to thrive and die between postnatal days 3 and 5. The skin is atrophic with reduced keratinocyte proliferation, but adhesion of keratinocytes to the basement membrane, differentiation and epidermal barrier are not altered. Perinatal lethality is due to severe progressive intestinal dysfunction, with severe inflammation and more than 80 % of the colonic epithelium detached by postnatal day 3 [11].
10.2.6 Kindlin-1 in Cancer
Several lines of evidence implicate kindlin-1 in cancer. First, increased mRNA levels of the kindlin-1 gene, FERMT1, were found in 60 % of lung and 70 % of colon cancers [32]. Second, FERMT1 is a TGF-β1 inducible gene, and its overexpression increases cell spreading and associates with epithelial to mesenchymal transition in HMEC (human mammary epithelial cells) [13]. Third, FERMT1 was identified as a gene associated with breast cancer lung metastasis [33], and there is evidence that kindlin-1 could also be an important mediator of lung metastasis in breast cancer and other cancer types metastasising to the lung [34].
10.3 Kindler Syndrome: Clinical and Molecular Features
10.3.1 Clinical Features
The Kindler syndrome (MIM# 173650), one of the four main epidermolysis bullosa types, is characterised by a mixed (intraepidermal and subepidermal) level of skin cleavage [5]. It is inherited in an autosomal recessive manner, and it is caused by mutations in FERMT1 (also known as KIND1, MIM# 607900), the gene encoding kindlin-1 [2]. To date, about 150 patients with Kindler syndrome due to FERMT1 mutations have been reported in the literature (https://grenada.lumc.nl/LOVD2/mendelian_genes/home.php?select_db=FERMT1).
The phenotype comprises skin blistering, photosensitivity and progressive poikiloderma with pronounced skin atrophy [35]. Skin blistering is usually present at birth and persists during childhood, but the tendency to develop blisters decreases with age. Most patients experience mild or no photosensitivity at all, but no objective evaluation of this feature is yet available. Skin atrophy of the dorsal aspects of the hands and feet strongly indicates the diagnosis of Kindler syndrome, and it occurs as early as the age of 2 years (Fig. 10.2a). Poikiloderma can be first recognised around the age of 10 years, first on sun-exposed areas and later disseminated to the entire integument. Sclerosing features of the hands and feet, such as webbing, sclerodermiform appearance of fingers or pseudoainhum, manifest in young adults with a significant variability (Fig. 10.2a).
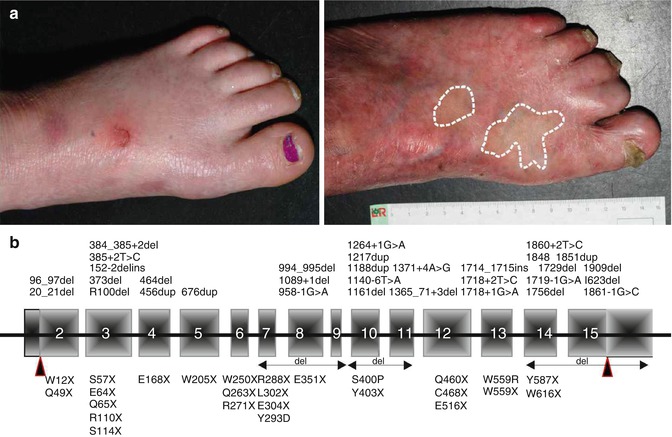
Fig. 10.2
Kindler syndrome: clinical and molecular genetic features. (a) Left feet of a 5-year-old girl with blisters and incipient skin atrophy (left) and of a 31-year-old man with normal-appearing revertant skin areas (circled), in contrast to the pronounced atrophy (right). (b) Schematic representation of FERMT1 with mutations reported in the literature
Mucosal involvement is very common in patients with Kindler syndrome. The oral mucosa is most frequently affected by fragility with mechanically induced bleeding and severe periodontitis. Many patients develop progressive dysphagia and oesophageal strictures requiring repeated dilatations. Anal, urogenital and ocular mucosa involvement is common, whereas intestinal involvement, i.e. colitis induced by epitheliolysis, was diagnosed in only a few cases [35].
In adults, the progressive phenotype is marked by poikiloderma and mucocutaneous scarring, leading to stenoses of the oesophagus and genitourinary tract, ectropion and webbing of fingers (Fig. 10.2a). Squamous cell carcinomas on the extremities, lips or oral mucosa, sometimes with severe aggressive course, have been reported [35, 36].
10.3.2 Disseminated Pattern of Revertant Mosaicism in Kindler Syndrome
Spontaneous gene repair, also called revertant mosaicism, has been documented in several genetic disorders involving organs that undergo self-regeneration, including the skin [37]. This phenomenon has been reported in all types of epidermolysis bullosa [37]. Notably, Kindler syndrome patients with FERMT1 duplication mutations demonstrate a particular, disseminated pattern of revertant mosaicism [38, 39] (Fig. 10.2a). Back mutations through slipped mispairing in direct nucleotide repeats were disclosed in all investigated revertant skin spots from two patients [38]. The sequence around the mutations demonstrated high propensity to mutations, favouring both microinsertions and deletions. Additionally, in some revertant patches mitotic recombination generated areas with homozygous normal keratinocytes. Restoration of kindlin-1 expression led to clinically and structurally normal skin regarding epidermal stratification and proliferation, as well as dermal-epidermal junction morphology. Since loss of kindlin-1 severely impairs keratinocyte proliferation, revertant cells have a selective advantage that allows their clonal expansion and, consequently, the improvement of the skin condition [38].
10.3.3 Diagnosis of Kindler Syndrome and Spectrum of FERMT1 Mutations
Morphological analysis of a skin biopsy is the first diagnostic procedure performed. The indirect immunofluorescence and transmission electron microscopy findings of irregular, branched and interrupted basement membrane are indicative of Kindler syndrome and justify mutation analysis, which represents the gold standard of Kindler syndrome diagnosis. FERMT1 is located on 20p21 and consists of 15 exons. Most of the about 60 mutations reported so far are predicted to lead to premature termination of translation and to loss of the kindlin-1 protein or of its function (https://grenada.lumc.nl/LOVD2/mendelian_genes/home.php?select_db=FERMT1) (Fig. 10.2b).
10.3.4 Kindlin-1 in the Pathogenesis of Kindler Syndrome
Insights into kindlin-1 biology should enhance our understanding of the pathogenesis of Kindler syndrome and represent a prerequisite for specific, rational therapeutic approaches.
In Kindler syndrome skin disorganised keratinocytes lose their proper architecture, polarisation and the boundary to the dermis [12]. In vitro loss of kindlin-1 is associated with abnormal cell shape, modification of the cortical actin network and increased plasticity of the plasma membrane, and, functionally, cell adhesion, spreading and directed motility are perturbed [9, 11, 12, 40]. These deficits stem from impaired functions of focal adhesions and related signalling pathways, with downstream effectors like Rho GTPases [20]. At the molecular level, expression of several proteins associated with cell-matrix and cell-cell adhesion, such as α6β4 integrin, collagen XVII, E-cadherin and desmoglein-3, is strongly reduced, whereas laminin 332 is synthesised in larger amounts than in normal keratinocytes. In contrast, mesenchymal markers such as vimentin and fibronectin are increased in keratinocytes lacking kindlin-1, suggesting the importance for maintenance of an epithelial phenotype [40]. A particular characteristic of Kindler syndrome is epidermal atrophy associated with minimal proliferation of basal keratinocytes and strongly impaired survival of keratinocytes in culture [11, 12, 20]. This feature might be related to β1 integrin subunit functions, but there has been no direct evidence to support this hypothesis.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


