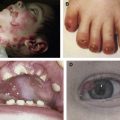
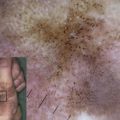
Kindler syndrome (MIM173650) is an autosomal recessive genodermatosis characterized by poikiloderma, trauma-induced skin blistering, mucosal inflammation, and photosensitivity. Loss-of-function mutations in the FERMT1 gene are the cause of Kindler syndrome. Kindler syndrome is categorized as a subtype of epidermolysis bullosa (EB). During infancy and childhood, there is clinical overlap between Kindler syndrome and dystrophic EB. Unlike other forms of EB, Kindler syndrome is characterized by impaired actin cytoskeleton-extracellular matrix interactions and a variable plane of blister formation at or close to the dermal-epidermal junction. This article reviews clinicopathologic and molecular features of Kindler syndrome and discusses patient management.
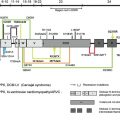
Understanding of blistering skin disorders involving the dermal-epidermal junction (DEJ) has improved considerably over the past 2 decades. This has been possible with the discovery of pathogenic mutations in genes encoding several structural components of cell–extracellular matrix (ECM) junctions in conjunction with the identification of autoantibodies targeting specific ECM proteins. Epidermolysis bullosa (EB) is the term used for the spectrum of inherited mechanobullous skin disorders that result from mutations in at least 13 different proteins’ involvement in epithelial or basement membrane integrity. Until the publication of the revised classification of EB in 2008, the molecular pathology of this condition was restricted to disorders that result from disruption of keratin intermediate filament-ECM anchorage. These skin fragility disorders include clinical subtypes of EB simplex, junctional EB, and dystrophic EB. In the latest classification of EB, however, Kindler syndrome (KS) (MIM173650) has been added as a further, specific form of EB. Trauma-induced blistering is common to KS and EB but individuals with KS soon develop progressive skin atrophy and poikiloderma. In addition, from a molecular viewpoint, KS differs from the other types of EB in that it results from disruption of the actin cytoskeleton-ECM anchorage network. The gene implicated in KS is FERMT1 , which encodes fermitin family homolog 1 (FFH1) protein, a focal adhesion protein that links the actin cytoskeleton with the underlying ECM. In the skin, FFH1 is predominantly expressed in the basal keratinocyte layer close to the DEJ. In cultured keratinocytes, it is present at focal adhesions where it mediates integrin-dependent ECM interactions. This article reviews the clinical and pathologic features of KS and provides an overview on patient management.
Background to Kindler syndrome
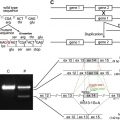
In 1954, Theresa Kindler reported the case of a 14-year old white girl who had congenital acral trauma-induced blistering and mottled pigmentation on her cheeks. This individual also burned easily in the sun and developed progressive skin atrophy. These features were suggestive of a subtype of EB and a form of inherited poikiloderma, but the precise nature of the disorder and the inheritance pattern, was unclear. In 1971, Weary and colleagues reported a family with overlapping clinical features. The pattern of inheritance was autosomal dominant but the clinical features were variable (vesicopustules, eczema, poikiloderma, and acral keratotic papules). The partial similarities between the Weary pedigree and the Kindler case report led to the introduction of the term, Weary-Kindler syndrome , and a further similar autosomal dominant pedigree was reported by Larregue and colleagues in 1981. Evidence for a distinct recessive disease, however, that more closely resembled the original Kindler case was published in 1985 and several other cases of unequivocal autosomal recessive inheritance followed. In recent years, characterization of the molecular basis of several genodermatoses that partly resemble KS has provided further evidence that it is a distinct disorder. The mechanobullous disease, dystrophic EB, has been shown to result from mutations in the type VII collagen gene, COL7A1 . Likewise, KS does not map to the Rothmund-Thomson syndrome gene, RECQL4, on 8q24.3 or other known DNA helicase genes. Another genodermatosis that overlaps with KS, EB simplex with mottled pigmentation, has been found to result from a specific mutation, p.Pro25Leu, in the cytokeratin 5 gene ( KRT5 ), thus also distinguishing this genodermatosis from true KS.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


