Fig. 3.1
Skin and keratin structures. (a) Schematic structure of the skin, epidermal layers (right side), and major site of expression of epidermal keratins (left side); (b) schematic structure of epidermal keratin protein with domain distribution. Regions most often affected by keratin mutations are marked in orange
Due to its constant exposure to the external environment, the epidermis has to cope with various forms of stresses such as mechanical friction and thermal injury [2]. Keratins form a highly organized spatial network of filaments spanning epithelial cell cytoplasm; they connect the cell membrane, to which they are attached at desmosomal and hemidesmosomal junctions, to the nuclear membrane and thereby play a major role in maintaining epidermal integrity, conferring to KC the ability to resist environmental strain [3].
Keratins are the most abundant structural proteins in the cytoplasm of epithelial cells; they belong to the intermediate filament protein family, a group of more than 70 distinct molecules, which are expressed in a tissue-specific manner [4]. Intermediate filaments are rod-shaped molecules characterized by a diameter of 10 nm, which is intermediate between the sizes of the two other major constituents of epithelial cell cytoskeleton, microtubules (25 nm), and microfilaments (8 nm). All keratins share a similar structure consisting in a central α-helical rod and two flanking non-helical (head and tail) domains. Two extremely well-conserved sequences are found at the beginning and at the end of the central rod domain, which are termed helix initiation and helix termination motifs (Fig. 3.1b).
The availability of a complete sequence of the human genome has led to the identification of more than 50 different keratin genes, which are classified as type I (acidic) and type II (basic) keratins and are organized in two genomic clusters on 17q21.2 and 12q13.3, respectively (with KRT18, encoding the type I keratin 18, being unusually located on chromosome 12). A novel nomenclature has recently been introduced to accommodate the new keratins [5]. Keratins are usually expressed and function as pairs of type I and type II keratins; the helix initiation and helix termination motifs play a crucial role in the process leading to the formation of these coiled-coil obligate heterodimers [4]. In addition to the large number of keratin genes, the promiscuity with which type I and II keratin proteins pair up and polymerize with one another [6] generates a tremendous potential for diversity in living cells.
An astounding two-thirds of known keratin genes are expressed in skin alone. Keratin gene expression pattern is tightly regulated and reflects the state of differentiation of KC [7]. Basal cell keratinocytes mostly express KRT5 and KRT14; suprabasal cells express KRT1 and KRT10, as well as KRT2 in the uppermost epidermal layers. In addition some keratins are expressed in the epidermis at specific anatomical locations only (e.g., KRT9 in the palmoplantar skin; KRT6b and KRT17 in the follicular epithelium) or under specific conditions (e.g., KRT6a and KRT16 during wound healing). Evidence suggests that although keratin proteins do not affect directly the execution of the differentiation program, the existence of a specific complement of keratin proteins has an important role in maintaining the normal cytoarchitecture and cellular functions in differentiated or specialized epithelial states. Keratin function involves a myriad of interactions with proteins responsible for their cross-linking (e.g., transglutaminase 1), polymerization (e.g., filaggrin), or proper attachment to the cell membrane (e.g., desmoplakin).
Although keratins are often considered as prototypic structural proteins, a steadily growing body of evidence implicates them in a number of nonstructural regulatory processes of importance, including protein synthesis [8], cell migration [9], and apoptosis [10].
3.2 Epidermolysis Bullosa Simplex (EBS)
3.2.1 History
EBS was originally described in 1898 by Hallopeau, following the original description of a congenital blistering disease by von Hebra, in 1870. It was not until the development of the electron microscope and its application to the skin that, in 1982, Anton-Lamprecht and Schnyder suggested that a defect in the function of keratins might cause EBS [11]. Subsequently, Akemi Ishida-Yamamoto and colleagues in Robin Eady’s group in London [12] provided important evidence that led to the genetic discoveries by Ervin Epstein’s group [13] and a collaboration between David Woodley, Amy Paller, and Elaine Fuchs [14] to demonstrate the involvement of the keratin 5 and 14 genes in the first human keratinopathy [15].
3.2.2 Epidemiology
The exact prevalence of EBS has been difficult to establish because a significant number of the milder cases are misdiagnosed as eczema or fungal infections. Various studies estimate the prevalence at about 6–30/106 live births [16–18]. In western countries, where consanguinity is rare, around 75–85 % of all EB cases are affected with EB simplex, inherited in an autosomal dominant fashion. However, in countries where consanguinity is more common such as Middle Eastern countries, autosomal recessive EB is more common, with EBS comprising only 50% of all EB cases [19].
3.2.3 Classification
A recent consensus classification refers to four main EB types according to the location of blister formation [20]: intraepidermal (epidermolytic, EBS), intralamina lucida (junctional EB, JBH), sublamina densa (dystrophic EB, DEB), and Kindler syndrome. According to this new classification scheme, EBS encompasses today both classical types of EBS, resulting from blister formation throughout the basal cell layer (EBS—basal type) as well as rarer disorders associated with suprabasal blister formation (EBS—suprabasal type, referred to in this chapter as EBS-SB). Subclassification of the various EBS types is based on the extent of skin involvement, mode of inheritance, and the mutated genes (Table 3.1).
Table 3.1
EBS-associated genes and phenotypes
Genes | Protein | Disease | OMIM |
|---|---|---|---|
KRT5 | Keratin 5 | EBS, Dowling-Meara type | 131760 |
EBS, other generalized (previously, Koebner type) | 131900 | ||
EBS, localized (previously EBS, Weber-Cockayne) | 131800 | ||
Epidermolysis bullosa simplex with mottled pigmentation | 131960 | ||
Epidermolysis bullosa simplex with migratory circinate erythema | 609352 | ||
Dowling-Degos disease | 179850 | ||
KRT14 | Keratin 14 | EBS, Dowling-Meara type | 131760 |
EBS, other generalized (previously, Koebner type) | 131900 | ||
EBS, localized (previously EBS, Weber-Cockayne) | 131800 | ||
Autosomal recessive EBS | 601001 | ||
Dermatopathia pigmentosa reticularis | 125595 | ||
Naegeli-Franceschetti-Jadassohn syndrome | 161000 | ||
PLEC1 | Plectin | EBS with muscular dystrophy | 226670 |
EBS Ogna type | 131950 | ||
Lethal EBS | 612138 | ||
EBS with pyloric atresia | |||
ITGB4 | Integrin β[beta]4 | Epidermolysis bullosa simplex junctionalis with pyloric atresia | 226730 |
EBS, localized (previously EBS, Weber-Cockayne) | 131800 | ||
COL17A1 | Collagen type XVII | EBS, other generalized (previously, Koebner type) | 131900 |
DSP | Desmoplakin | Epidermolysis bullosa, lethal acantholytic | 609638 |
PKP1 | Plakophilin-1 | Ectodermal dysplasia/skin fragility syndrome | 604536 |
DST | Dystonin (BPAG1-e) epithelial isoform of bullous pemphigoid antigen 1 | EBS, autosomal recessive with neurologic symptoms |
3.2.4 Pathogenesis
3.2.4.1 EBS Caused by Mutations in Keratin Genes
Most cases of EBS are caused by heterozygous missense mutations in KRT5 and KRT14, encoding keratins mostly expressed in the epidermal basal layer [7, 21]. While the overwhelming proportion of EBS cases in the Western world are inherited in an autosomal dominant (a.d.) fashion, in countries where marriage within the same community/family is common, recessive cases are more prevalent. For example, in the Middle East, approximately 30 % of the cases are caused by bi-allelic recessive mutations in KRT14 [19, 22]. More complex patterns of inheritance may exist, as recently exemplified by a report on digenic inheritance in a child affected by a generalized form of EBS [23]. Mutations in other genes encoding basement membrane zone proteins have also been shown to account for a minority of EBS cases, as will be discussed later.
Most KRT5 and KRT14 mutations have been shown to disrupt the central alpha-helical segment of these keratin molecules, thereby compromising the structure and function of the cell cytoskeleton which becomes unable to accommodate even small amount of mechanical stress. As a consequence of keratin cytoskeleton dysfunction, the basal cell layer is prone to cytolysis when exposed to friction forces. At the ultrastructural level, keratin abnormal function translates into cell vacuolization, keratin filament clumping, and blister formation [15]. Phenotype-genotype analysis revealed that mutations affecting conserved areas at the beginning and end of the central rod segment are usually associated with a more severe phenotype (EBS Dowling-Meara, EBS-DM) than mutations affecting less conserved areas of the keratin molecules (EBS localized, EBS-loc) [24–26], although many exceptions to this rule have been reported [19, 22]. In addition, the nature of the amino acid substitution and not only its location can influence the severity of the disease as well [27, 28].
Most EBS-causing mutations exert a dominant-negative effect, namely, the mutant molecules interfere with the function of the normal keratins encoded by the wild-type allele. This situation has direct implications for the design of genetic therapies for EBS. Indeed, introduction of a wild-type allele is unlikely to benefit EBS patients; instead, effective therapies for EBS should be aimed at eliminating the deleterious keratin molecules encoded by the mutant allele (see below).
It should be noted that EBS phenotype usually evolves over time and generally show improvement as affected individuals get older. Reduced expression of mutant keratin genes and compensatory overexpression of keratins usually weakly expressed in the basal cell layers, such as KRT15 [29], have been invoked to explain this phenomenon. Somatic genetic events may also modify the course of the disease. Revertant mosaicism refers to a situation where a second mutation attenuates or abolishes the deleterious effect of the original mutation in certain areas of the skin. This phenomenon has been reported in a number of patients with EBS and may actually be more common than previously suspected [30, 31]. The phenotypic manifestations of EBS-causing keratin mutations can also be influenced by apparently silent sequence alterations [32]. Genetic background is also important as exemplified by the fact that phenotype-genotype correlations differ across populations and families [19] and by the fact keratin mutations are phenotypically expressed in a strain-dependent fashion in mice [33]. Finally, nongenetic factors may also determine the way a given sequence alteration manifests as illustrated by a transient EBS-like phenotype associated with bexarotene treatment in the presence of an otherwise silent polymorphism in KRT5 [34].
As reviewed so far, a wealth of evidence supports the view that structural fragility of basal keratinocytes accounts for the generation of skin blisters in individuals with EBS, reflecting a loss of epidermal integrity following incipient mechanical trauma [35]. Nevertheless, other mechanisms may also contribute to the disease phenotype. For example, recent data implicate excessive apoptotic activity, possibly induced by keratin clumps, and upregulation of the inflammatory response in the pathogenesis of EBS [36, 37]. In addition, some keratin mutations may affect the cytoskeletal dynamics or interfere with normal keratin post-translational modification [38, 39]. For instance, the presence of cytoplasmic aggregates containing mispolymerized mutant keratin proteins, a defining characteristic of EBS-DM, may contribute to the pathophysiology at the cellular and tissue levels. Conceivably, the failure of misfolded protein response to resolve these aggregates may lead to cellular and tissue stress [40, 41]. Also, gene expression analysis of an EBS-DM cell line has demonstrated upregulation of genes controlling epidermal development, migration, apoptosis, and wound healing as a molecular consequence of a KRT14 mutation [42].
Transgenic mouse models have also revealed several novel functions for keratin proteins in skin epithelia [43] which may all be of relevance to the pathogenesis of EBS, including regulation of cell and tissue growth in the epidermis and hair follicle [10, 44] and promotion of keratinocyte proliferation correlating with the expression of proinflammatory and/or mitogenic cytokines and chemokines in keratinocytes [45]. A recent genome-wide screen has implicated KRT6, KRT16, and KRT17 in a genetic network defining the key interrelationship between barrier function, inflammation, and tumor susceptibility in mouse skin [46]. In keeping with the theme of inflammation and chemokines, Roth et al. [47] have reported an increased density of Langerhans cells in mice null for KRT5 and in individuals with EBS resulting from mutations at the KRT5 locus. This phenomenon correlates with an upregulation in chemokines (e.g., CCL2, CCL19, and CCL20) known to attract Langerhans cell precursors to the skin and otherwise adds to the emerging concept that keratins may act as key immune modulators in the skin [45].
It seems that epidermal fragility is almost exclusively associated with defective function of the conserved central regions of keratin molecules since skin blistering is unusual in disorders resulting from mutations affecting the head or tail domain of keratins. For example, in Dowling-Degos disease (DDD; MIM179850) resulting from mutations in the KRT5 gene region encoding the protein head domain [48], blistering is not observed (although acantholysis is often seen [49]); in contrast, melanosome transport and epithelia growth are abnormal, resulting in reticulate hyperpigmentation of the flexures, comedo-like lesions on the neck, and pitted perioral acneiform scars [50]. In Naegeli-Franceschetti-Jadassohn syndrome (NFJS; 161000) and dermatopathia pigmentosa reticularis (MIM125595), which are caused by mutations affecting the head domain of KRT14 [51], blisters are very unusual; instead the patients display reticulate hyperpigmentation and lack dermatoglyphics [52], due to deranged regulation of apoptotic activity in the basal cell layer [53]. EBS with mottled pigmentation (MIM131960) is characterized by skin blistering associated with reticulate skin pigmentation and is often caused by a recurrent missense mutation (p.P24L) affecting KRT5 head domain [54]. Altogether, these data suggest a role for the non-helical domain of basal keratins in regulating skin pigmentation. No mechanism has been established to account for these pigmentation phenotypes, although there is preliminary evidence pointing to an interaction between KRT5 and components of microtubule-dependent motors, which are involved in melanin pigment transport [48, 55].
3.2.4.2 EBS Caused by Mutations in Nonkeratin Genes
A number of subtypes of EBS are not caused by mutations in keratin genes per se but rather result from defective function of molecules associated with keratins. These can be subclassified as basal vs. suprabasal forms of EBS.
Mutations affecting intracellular components of the hemidesmosomes result in basal EBS, underscoring the interdependency between hemidesmosomal junction and epidermal cell cytoskeleton functions. For example, mutations in the genes encoding integrin β[beta]4 and collagen type XVII have been found to cause EBS [56, 57]. A new form of autosomal recessive (AR) EBS was recently described as due to biallelic mutations in the DST gene that encodes the coiled-coil domain of the epithelial isoform of bullous pemphigoid antigen 1, BPAG1-e (also known as BP230) [58, 59]. A number of relatively rare forms of EBS associated with suprabasal blistering are now recognized (EBS-SB). Ectodermal dysplasia with skin fragility (EDSF) (MIM604536) is caused by mutations in PKP1, encoding plakophilin 1, a component of the desmosomal plaque [60–62]. Keratin intermediate filaments binding to the desmosomal plaque, at least in lower suprabasal epidermal cells, are critically dependent upon normal PKF1 function [63], which may explain the common occurrence of blistering in EBS and in EDSF. Lethal acantholytic epidermolysis bullosa (LAEB) is an AR disorder caused by mutations in DSP encoding the desmosomal protein, desmoplakin (DSP). LAEB-causing mutations result in truncated DSP polypeptides lacking the tail domain of the protein, which play a pivotal role in binding to keratin filaments [64]. Histology shows suprabasal clefting and acantholysis throughout the spinous layer, mimicking pemphigus. A second form of lethal congenital EB is caused by a homozygous nonsense mutation in JUP encoding plakoglobin [65]; the aforesaid is a constituent of desmosomes and adherens junctions, and the complete loss of the protein affects expression and distribution of desmosomal components which highlights the fundamental role of plakoglobin in epidermal cohesion.
3.2.5 Clinical Manifestations
EBS age of onset is very variable, with the most severe cases manifesting at birth and mild cases first appearing during the second or third decade of life only. All forms of EBS manifest with blistering of the skin, usually induced by exposure to mechanical friction or trauma (Fig. 3.2a–c). High ambient temperatures and sweating are also aggravating factors. EBS is associated in severe cases with palmoplantar keratoderma (thickening of the skin) (Fig. 3.2e), nail dystrophy, and mucosal tissue involvement.
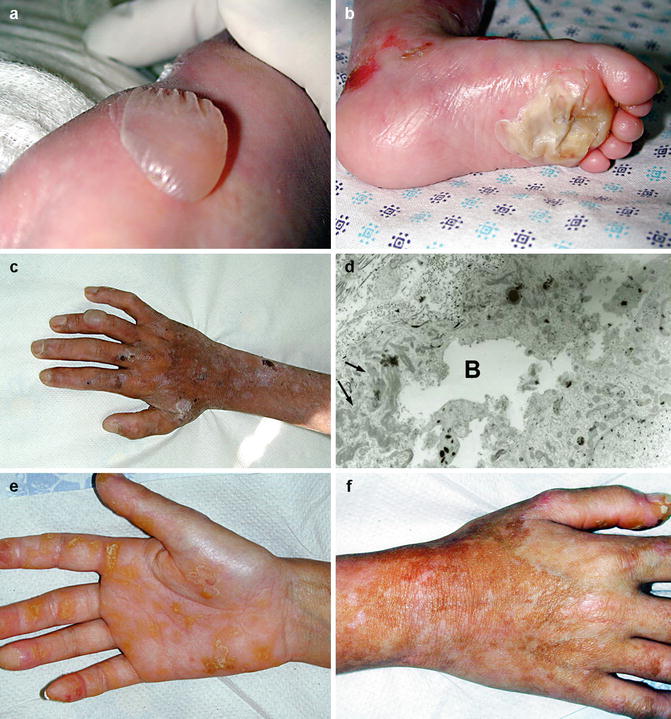
Fig. 3.2
Clinical features in EBS. Subepidermal blistering (a) over the left thigh of a young child with EBS, Dowling-Meara type, (b) on the plantar surface of an EBS patient, and (c) on the dorsum of adult with EBS, Koebner type; (d) electron micrograph demonstrating blister formation through the basal cell layer (b) as well as keratin filament clumping in the basal cell cytoplasm (arrows); (e) palmar hyperkeratosis in a patient with EBS, Dowling-Meara type; (f) mottled hyperpigmentation in EBS-MP
Severe skin blistering is associated with marked morbidity (pain, infections, fluid and electrolyte imbalance, malnutrition, anemia) as well as elevated mortality [66]. More recently reported complicating conditions include EB nevi and malignancies. EB nevi have been reported in all major forms of EB and may simulate clinically and dermoscopically melanoma (although no malignant transformation of these lesions has been reported and they often disappear spontaneously) [67, 68]. Severe forms of EBS are associated with an increased risk for skin cancer [69] and death [66]. Aside from the clinical manifestations described above, EB has a clinical and socioeconomic impact on patients and their families; therefore, psychological support is vital for patients and their caretakers [69, 70].
Several unusual EBS variants deserve a special mention:
Recessive EBS (MIM601001) can be caused by either missense or nonsense mutations in KRT14 resulting in loss of function rather than a dominant-negative effect [19]. In the case of null mutations, the disease can easily be diagnosed by demonstrating the absence of mature tonofilaments (e.g., keratin bundles) on electron microscopy or lack of immunostaining for KRT14 on paraffin-embedded sections.
As mentioned above, EBS with mottled pigmentation (MIM131960) is characterized by skin blistering, reticulate skin pigmentation, keratoderma, and nail dystrophy (Fig. 3.2f). This subtype of EBS has been found to be strongly associated with a missense mutation (p.P24L) affecting KRT5 head domain [54], although the same phenotype has also been reported with other mutations in KRT5 and KRT14 [71].
Epidermolysis bullosa simplex with migratory circinate erythema (MIM609352) is characterized by the occurrence of vesicles on the background of a migratory circinate erythema (Fig. 3.2c). The lesions often heal with brown pigmentation but no scarring. The disease seems to be specifically caused by a recurrent frameshift mutation affecting the structure of KRT5 tail domain [72]. The reason for this peculiar association is still elusive.
Mutations in PLEC1, encoding plectin, a large molecule that is part of the hemidesmosome and is known to interact with basal keratins, were found to cause a variety of EBS subtypes [73], including EBS with muscular dystrophy (MIM226677), which may also present with cardiomyopathy [74]; EBS Ogna type (MIM131950) characterized by hemorrhagic blistering; lethal EBS; and EBS with pyloric atresia (MIM612138), which is also caused by mutations affecting the α[alpha]6β[beta]4 integrin receptor, another component of the hemidesmosomal plaque [15].
Suprabasal EBS is a recently recognized subtype of EBS and includes disorders already mentioned above such as ectodermal dysplasia skin fragility syndrome (EDSF), featuring suprabasal blistering, nail dystrophy, palmoplantar keratoderma, and alopecia [60–62] as well as lethal acantholytic EB, which manifests with skin fragility, complete disruption of the epidermal barrier, universal alopecia, neonatal teeth, and nail loss [75].
3.2.6 Diagnosis
The diagnosis of EBS can be established by the use of three main techniques: immunofluorescence mapping (IFM), transmission electron microscopy (EM), and mutation analysis [20]. Transmission EM has been considered as gold standard in the diagnosis of EB until recently, allowing for visualization of the skin ultrastructure and revealing the exact location of blister formation as well as keratin filament clumping in Dowling-Meara-type EBS (Fig. 3.2d) [24, 25, 76]. The technique of IFM detects structural proteins in the basement membrane zone by using specific antibodies. It allows localization of the level of blistering and detects reduced or absent protein expression. In contrast with EM, IFM is readily available at most medical institutions. Also it was reported to be more sensitive and specific than EM [20, 77]. In addition, it allows for the detection of nonspecific but very sensitive histopathological markers of keratinopathies, such as dyskeratosis [78].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


