Subtype
Onset
Clinical features
Gene
Inheritance
IF
TEM
JEB, generalized severe
Birth
Lethal in childhood; extensive generalized blistering; dyspnea; anemia; failure to thrive; nail loss and abnormalities; enamel hypoplasia
LAMB3/LAMC2/LAMA3
AR
LM-332 absent or strongly reduced
Lamina lucida split; HD reduced number and hypoplastic; absent subbasal dense plates
JEB, intermediate, generalized
Birth
Generalized blistering; enamel hypoplasia; nail abnormalities; primary hair (incomplete) alopecia; secondary hair absent; atrophic scarring; corneal blistering
COL17A1/LAMB3/LAMC2/LAMA3/ITGB4
AR
C17 absent; LM-332 reduced or normal; β4 reduced or normal
Lamina lucida split; HD normal or HD reduced number and hypoplastic
JEB, localized
Birth
Blistering hands and feet; enamel hypoplasia; nail abnormalities; normal primary hair; sparse secondary hair
COL17A1/LAMB3/LAMA3/ITGB4/(LAMC2?)
AR
C17 reduced; LM-332 reduced or normal; β4 reduced or normal
Lamina lucida split; HD normal or HD reduced number and hypoplastic
JEB with pyloric atresia
Birth
Frequently lethal; pyloric atresia; aplasia cutis; enamel hypoplasia; nail abnormalities; urinary tract stenosis
ITGB4/ITGA6
AR
α6β4 absent or reduced
HD reduced number and hypoplastic
JEB, late onset
Childhood-young adulthood
Blistering hands and feet; hyperhidrosis; loss of dermatoglyphs; skin atrophy; enamel hypoplasia; nail abnormalities
COL17A1
AR
C17 staining normal in intensity but altered distribution
Electron dense cloudy broadening of (sub)lamina densa (zone)
LOC syndrome
Birth
Hoarse cry after birth; chronic mucosal, laryngeal, and ocular granulation tissue; enamel hypoplasia
LAMA3
AR
LM-332 stains in normal intensity and normal distribution
Normal number of HD; some HD plaques are small, lacking normal subbasal dense plates; reduced number of anchoring filaments
37.2 Junctional Epidermolysis Bullosa, Generalized Intermediate
Generalized intermediate JEB (Fig. 37.1), formerly referred to as non-Herlitz JEB or generalized atrophic benign epidermolysis bullosa (GABEB), was first described in 1976 by Hashimoto, Schnyder, and Anton-Lamprecht and named Disentis type (MIM 226650) after the Swiss valley where the patient lived [28]. The term GABEB with atrophic skin and alopecia was introduced in 1982 by Hintner and Wolf but is now regarded as synonymous with JEB-gi [29]. The clinical features of this form of EB are continuous blistering from birth, cigarette paper-like atrophic hypopigmented skin, absent scarring and milia at sites of recurrent blistering, mild mucous membrane involvement, dystrophic nails and dentition, and atypical alopecia with significant scalp atrophy, partial absence of eyelashes and eyebrows, and absent pubic and axillary hair.
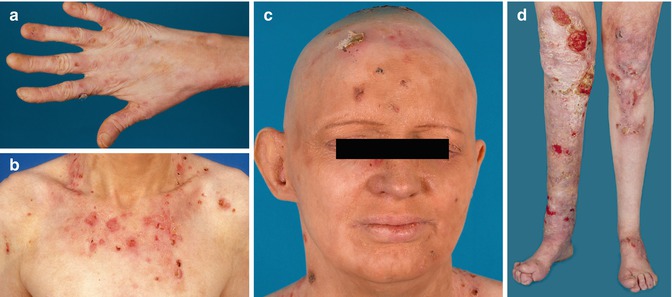
Fig. 37.1
Generalized intermediate junctional epidermolysis bullosa phenotype (EB 117–01) with blistering on hands (a) and trunk (b), absent fingernails (a), and universal alopecia (c). At the age of 41 years, she developed a squamous cell carcinoma on the right knee and lower leg (d) (From Pasmooij et al. [11])
In localized JEB (Fig. 37.2), blistering is confined to hands, lower legs, and face, the skin is normally pigmented, nail dystrophy is absent or very mild, and secondary hair is sparse, while scalp hair is normal.
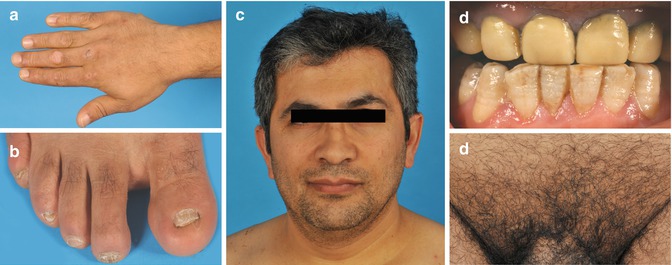
Fig. 37.2
Localized junctional epidermolysis bullosa (EB 168–01) with blistering limited to hands (a) and feet (b) and abnormal nails (b), but normal hair pattern of scalp, beard (c), and pubic region (e). Teeth enamel showed dental pitting (d) (From Pasmooij et al. [11])
37.2.1 Revertant Mosaicism
Patients with JEB-gi due to COL17A1 and LAMB3 mutations frequently have revertant mosaicism: previously described in as many as 35 % of cases [30], a recent series of patients with COL17A1-mutant JEB-gi have all been shown to have areas of revertant mosaicism recognized by skin hyperpigmentation [31].
Revertant mosaicism may be unnoticed in localized JEB localized [31], possibly because revertant patches do not stand out against the surrounding mildly affected mutant skin or because basal keratinocytes with revertant C17 do not have a growth advantage against those with reduced C17 in localized JEB.
37.2.2 Teeth
All JEB-gi patients have abnormalities of primary and secondary dentition. LM-332 and C17 are crucial in ameloblast differentiation and enamel formation, resulting in enamel defects of the entire dentition, consisting of hypoplasia, pitting, roughness, and thinning or furrowing of enamel in all patients with JEB-gi [32]. COL17A1 glycine substitutions may have a dominant-negative effect on dentition with enamel hypoplasia and pitting in carriers with [33] or without [34] skin blistering. In one study, carriers of mutations in the C17 gene displayed enamel hypoplasia [35], whereas in another it was absent [13].
Yuen et al. reported two related heterozygous carriers of the novel LAMA3 deletion c.488delG who did not suffer from skin blistering, but showed dental enamel defects, consisting of roughness and pits, which led to a higher susceptibility of caries in both [36]. As the mutation c.488delG creates a frameshift resulting in a PTC and mRNA decay, and as alternatively spliced cDNA products could not be found in cDNA analysis, they hypothesized that haploinsufficiency of laminin α3 is the cause of the enamel defects in these carriers and that in the complex process of enamel formation in which LM-332 plays such a vital role, there is no compensation for the loss of one LAMA3 allele, whereas this mechanism exists in the skin. However, it remains notable that enamel defects have not been reported in carriers of LAMB3 or LAMC2 null mutations.
37.2.3 Wound Healing
Patients with LM-332-deficient JEB-gi may be clinically different from those with C17 or α6β4 deficiency due to the occurrence of persistent deep small ulcers [21, 37]. These patients suffer from impaired wound healing due to LM-332 deficiency, mostly caused by mutations in LAMA3. The predilection sites are palmoplantar and pretibial skin and on hips and buttocks. LM-332 plays an important role in the process of epidermal wound healing, where cell motility is necessary for wound closure [38]. Basal keratinocytes secrete unprocessed LM-332 around the wound edge. In its unprocessed form, the LG3 domain of the laminin α3 chain favors interaction with integrin α3β1 (α3β1). This interaction stimulates the mitogen-activated protein (MAP) kinase signaling cascade to promote epithelial cell migration and proliferation. Upon wound closure, the LG4–5 domain of the laminin α3 chain is cleaved by plasmin. This changes the favored interaction of LM-332 with α3β1 to an interaction with α6β4, after which hemidesmosomes are formed [38–46]. LM-332-α3β1 interaction enhances plasmin production, making the process self-regulatory with an inhibitory feedback loop [46, 47].
JEB-gi wounds have been successfully treated by simple punch grafting of autologous (mutant) skin [37].
37.2.4 Squamous Cell Carcinoma
The frequency of developing a cutaneous squamous cell carcinoma (SCC) among adult JEB patients in the Dutch center was 25 % (n = 7) [48]. In the literature, seven case reports of JEB complicated by SCC are published, bringing the total number of documented cases to 14 [49–56]. All patients were adults and classified as JEB-gi. The first SCC developed at an average age of 50 years (median 52 years, range 28–70 years). In nine patients, multiple primary SCCs occurred, with a total of 45 SCCs. The SCCs were most often located on the lower extremities, in areas of chronic blistering, long-standing erosions, or atrophic scarring. The treatment of first choice is surgical excision, although no guidelines for the excision margins are given. Three (21 %) patients developed metastases and died on average 8.9 years after diagnosis of the initial SCC. Because SCCs can develop in early adulthood, we recommend checking the entire skin of JEB patients from age 25. Although SCCs can look like normal wounds, suspicious lesions are chronic nonhealing ulcers or erosions, hypergranulation, induration, and hyperkeratotic, exophytic, verrucous nodules or plaques. Additional clues can be pain, a stinging or burning sensation, and noticeable changes in a lesion. Multiple biopsies of suspicious lesions should be taken to avoid missing malignancies.
The role of LM-332 seems to be more complicated in carcinogenesis. Cell migration and invasion are important stages in tumor progression and establishing metastasis. The basement membrane zone (BMZ) has traditionally been viewed as a protective barrier, and its proteolytic degradation by metalloproteinases merely as a way to facilitate tumor spread [57]. However, in a great diversity of cancers, expression of LM-332 is elevated and is considered as a predictive factor related to tumor invasiveness [58–61]. Considering the importance of (increased) expression of LM-332 in cell migration and carcinogenesis, it seems paradoxical that LM-332-deficient JEB-gi patients have an increased risk of developing invasive metastatic SCC, whereas they also suffer from an impaired wound healing. Further research in the tumor microenvironment of LM-332-deficient JEB-gi keratinocytes should be performed to gain more clarity on this subject.
37.3 Junctional Epidermolysis Bullosa with Pyloric Atresia
JEB with pyloric atresia (JEB-PA) (Fig. 37.3) is clinically characterized by generalized blistering, gastrointestinal atresia usually affecting the pylorus, but occasionally also other sites such as the duodenum or anus, aplasia cutis, and urinary tract stenosis associated with the complete or partial absence of integrin α6β4 (α6β4) [15, 16, 20]. In most cases, JEB-PA has a fatal outcome despite surgical correction of the pyloric atresia; however, some milder nonlethal cases have been described [16, 17]. Most patients with JEB-PA have mutations in ITGB4 (94.6 %), while a minority have mutations in ITGA6 (3.4 %) [62]. The genotype-phenotype correlation between lethal and nonlethal JEB-PA is not completely elucidated: it is hypothesized that the types of mutations (nonsense versus missense) as well as the specific sites at which they reside are responsible for the resultant phenotype [20].
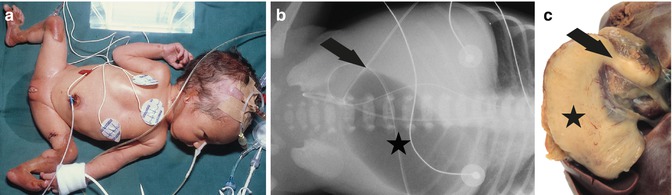
Fig. 37.3
Lethal junctional epidermolysis bullosa with pyloric atresia (EB 033–01) shows cutis aplasia of lower legs and ears (a). X-ray abdomen (b) reveals gastric bubble (asterisk) and airless intestines due to pyloric obstruction (arrow). Preparation of stomach (asterisk) at autopsy shows pyloric atresia (arrow) (c)
37.4 Rare Subtypes
37.4.1 Junctional Epidermolysis Bullosa of Late Onset
JEB of late onset (JEB-lo) (Fig. 37.4) is a very rare form of JEB, recently shown to arise from mutations in COL17A1 [63]. In this report, two siblings with JEB-lo suffered from blistering on the feet and around the toe- and fingernails starting at the age of 6 years. With age, blistering also occurred on the hands, nose, and oral mucosa, along with nail deformities, pretibial atrophic patches, palmoplantar hyperhidrosis, loss of dermatoglyphs, and amelogenesis imperfecta. However, the primary and secondary hair pattern was normal.
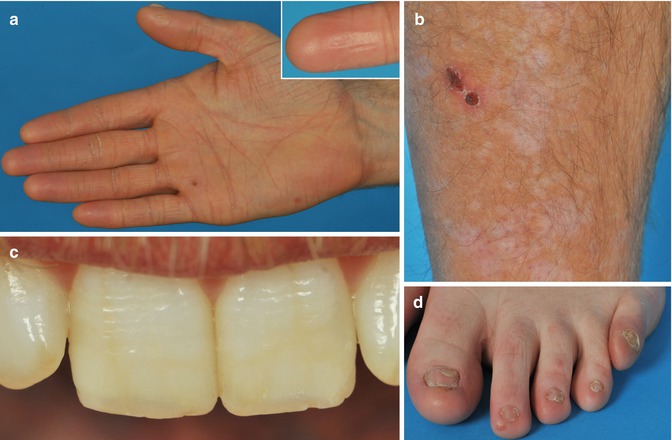
Fig. 37.4
Junctional epidermolysis bullosa of late onset (EB 054–01) shows loss of dermatoglyphs and waxy palmar hyperkeratosis (a), atrophic skin on the lower leg (b), transverse ridging and enamel pits of the teeth in patient (c), and abnormal nails (d) (From Yuen et al. [62])
IF antigen staining showed loss of the apical-lateral C17 staining and a broadened distribution of staining against the ectodomain of C17, LM-332, and type VII collagen. Mutation analysis of COL17A1 showed compound heterozygosity for a novel mutation c.1992_1995delGGGT and the known mutation c.3908G>A in both patients. The deletion c.1992_1995delGGGT results in a PTC and mRNA decay, leaving the patients functionally hemizygous for the missense mutation c.3908G>A (p.R1303Q). It is hypothesized the mutation p.R1303Q, located in the C17 ectodomain, results in a disturbed ligand binding that interferes with the normal deposition of extracellular matrix, explaining the broadened IF staining against LM-332 and type VII collagen, as the C17 ectodomain has an interaction with LM-332 and thus with type VII collagen.
In a previous study, a patient carrying the mutation p.R1303Q homozygously showed clinical symptoms matching JEB-lo [9]. This supports a hypothesis that p.R1303Q and other missense mutations in the NC4 domain of C17 are specific for JEB-lo. To date, no other missense mutations in the NC4 domain have been described.
37.5 Protein- and DNA-Based Diagnosis
Immunofluorescence antigen mapping (IF) is the major diagnostic tool for EB with a specificity and sensitivity of 100 and 90 %, respectively [64]. IF staining with antibodies to the C17 endodomain can distinguish between healthy subjects and all C17-related EB subtypes and carriers [65]. Apical-lateral C17 staining of basal keratinocytes is only present in healthy non-carrier subjects. Furthermore, C17 staining can be used to distinguish generalized from localized JEB-gi [13]. A total absence of C17 is seen in generalized JEB-gi and is caused by COL17A1 nonsense mutations, out-of-frame deletions or insertions, and deleterious glycine substitution missense mutations (Table 37.1) [13, 14, 66, 67]. In contrast, localized JEB is associated with a reduction of C17 along the epidermal BMZ due to COL17A1 missense mutations or small in-frame deletions or insertions [9, 13, 14, 68]. This indicates that mAbs against the C17 endodomain (1A8c, VK1–5) are the most sensitive in detecting reduced C17 production, as they only detect full-length C17, while mAbs against the C17 ectodomain detect both full-length C17 and shed C17 products.
In JEB generalized severe, total absence of staining to LM-332 with the antibody GB3 is almost always seen [69–71]. However, in a minority of cases, strongly reduced GB3 staining may be seen [5], a staining pattern also seen in JEB-gi cases. In these cases, further molecular analysis, multiple biopsies, and cDNA analysis should determine the definite diagnosis. For LM-332-deficient JEB, the distinction between JEB-gi and JEB-localized remains more obscure, with an overlap in the amount of GB3 staining seen. Although a (strongly) reduced GB3 staining is only seen in JEB-gi, a slightly reduced or normal GB3 staining can both be seen in JEB-gi and localized JEB. A third of JEB-gi cases show normal GB3 staining, with the consequence that no candidate gene can be found in these cases [65].
In JEB-PA patients, abnormal α6 and β4 staining is seen, indicating the causative protein, integrin α6β4. Also, all known ITGB4-associated JEB cases have shown reduced β4 staining compared to normal, making IF a useful diagnostic tool. Prognostically, it has been postulated that a total absence of either α6 or β4 staining is consistent with lethal JEB-PA and a reduced staining with nonlethal JEB-PA. This has been supported by an IF mapping study in 26 JEB-PA cases: most patients with a total absence of either α6 or β4 staining did not survive, although there was an exception to this rule [72]. However, if α6 and β4 were both reduced, no significant correlation was seen with survival. Completely absent staining for an epitope on the β4 endodomain may be caused by epitope skipping resulting in mild β4-associated localized JEB [73]. Ideally, therefore, multiple antibodies should be used in IF antigen mapping for the same polypeptide if absence of staining is found with any.
Related posts:
 Kindlin-1 and Its Role in Kindler Syndrome
Kindlin-1 and Its Role in Kindler Syndrome
 Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
 Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
 Living with Epidermolysis Bullosa: Reviewing the Impact on Individuals’ Quality of Life
Living with Epidermolysis Bullosa: Reviewing the Impact on Individuals’ Quality of Life
 How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


