Inherited Epidermolysis Bullosa: Introduction
|
Introduction
Inherited epidermolysis bullosa (EB), is a family of diseases, with the common feature of blistering in response to mild trauma. Patients with EB can show blistering in the form of small vesicles or larger bullae, which can occur on both the cutaneous surfaces as well as on the mucosal tissues. The fragility of the skin and mucosa and the traumatic production of painful blisters are what all EB cases share in common. However, the distribution of the involvement, the depth of blister formation, any associated extracutaneous involvement and the severity of the blistering process vary with the different EB subtypes and depend on the underlying heritable molecular defect. Different EB subtypes also vary in the way in which blistered areas heal. The wound repair responses are often abnormal and can eventuate into chronic erosions, hypertrophic granulation tissue, scarring, or even invasive carcinoma. While the milder EB subtypes are associated with a normal lifespan and little or no internal involvement, the most severe recessively inherited forms are mutilating, multiorgan disorders that threaten both the quality and length of life.
A number of early studies identified the major subtypes of EB. Studies of von Hebra1–3 were the first to distinguish pemphigus from inherited blistering and the term epidermolysis bullosa hereditaria was first suggested by Koebner.4 Hallopeau was the first to distinguish between simplex (nonscarring) and dystrophic (scarring) forms of the disease5 while Weber6 and Cockayne,7 Dowling and Meara8 and Koebner4 each described unique forms of epidermolysis bullosa simplex. Hoffman,9 Cockayne,10 Touraine,11 Pasini,12 and Bart13 provided much of the information about subtypes of dystrophic epidermolysis bullosa. Herlitz described epidermolysis bullosa letalis,14 which was later found to be a part of the third major category of epidermolysis bullosa: the junctional form. The application of electron microscopy toward diagnosis of epidermolysis bullosa led to the studies of Pearson15 and collaborators who classified the patients not only on the basis of clinical findings but also on the existence of ultrastructural changes. A comprehensive classification of epidermolysis bullosa based on a combination of ultrastructural and clinical findings was completed in an early landmark treatise by Gedde-Dahl.16 Recent major advances have led to the identification of protein and genetic abnormalities in most types of epidermolysis bullosa patients. These studies have led to an improved understanding of the biological basis of epidermolysis bullosa and, finally, a classification of epidermolysis bullosa based on genetic/protein defects, which provides a rational approach to specific molecular therapy.
Etiology and Pathogenesis
Epidermolysis bullosa arises from defects of attachment of basal keratinocytes to the underlying dermis. These defects can arise from inside the keratinocyte plasma membrane or extracellularly in the dermal–epidermal basement membrane zone (BMZ). Many tissues such as the skin and cornea, which are subjected to external disruptive forces, contain a complex BMZ composed of a group of specialized components that combine together to form anchoring complexes (Fig. 62-1). At the most superior aspect of the BMZ, keratin-containing intermediate filaments of the basal cell cytoskeleton insert upon electron dense condensations of the basal cell plasma membrane termed hemidesmosomes. Anchoring filaments span the lamina lucida connecting hemidesmosomes with the lamina densa and anchoring filaments. At the most inferior aspect of the BMZ, collagen VII-containing anchoring fibrils extend from the lamina densa into the papillary dermis and combine with the lamina densa and anchoring plaques, trapping interstitial collagen fibrils. Thus, the cutaneous BMZ connects the extensive basal cell cytoskeletal network with the abundant network of interstitial collagen fibrils in the dermis.17,18
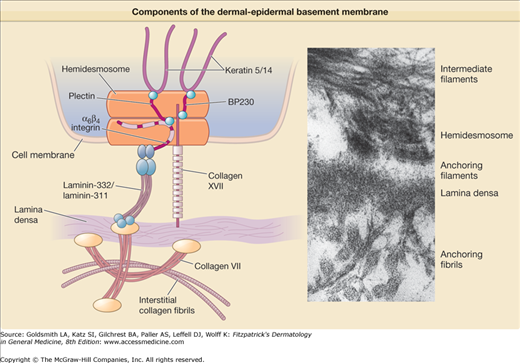
Keratins are obligate heteropolymers that are composed of pairs of acidic and basic monomers. The keratin pair 5 and 14 assembles together to form the extensive intermediate filament network of the basal cell cytoskeleton.19 Keratins contain a central α-helical rod with several nonhelical interruptions as well as nonhelical carboxyl and amino-terminal regions. The regions of highest conservation between the keratins are located on the ends of the keratin rod in the helix boundary motifs. Extensive mutagenesis studies suggest that helical regions near the ends of the central rod are important in keratin filament elongation, whereas the nonhelical domains may be important in forming lateral associations.20
Hemidesmosomes contain intracellular proteins, including plectin and BP230. Plectin is a 500-kDa protein, which acts as an intermediate filament-binding protein. It is possible that plectin also interacts with microfilaments, as plectin contains a domain with similarity to the actin-binding domain of spectrin.21,22 BP230, also known as BPAG1, is a 230-kDa protein which has homology both to desmoplakin23 and to plectin. Several splicing variants of BP230 are of vital importance in the nervous system.24–26 BP230 localizes to a region referred to as the inner plate on the cytoplasmic surface of the hemidesmosome and like plectin, functions in the connection between hemidesmosomes and intermediate filaments. BP230 negative transgenic mice lack a hemidesmosomal inner plate and the connection between hemidesmosomes and intermediate filaments is severed, creating a cytoplasmic zone of mechanical fragility just above the hemidesmosomes.
Hemidesmosomes also contain the transmembrane proteins collagen XVII (also termed BPAG2 and BP180)27 and α6β4 integrin.28 The cytoplasmic portions of these molecules make up part of the hemidesmosome dense plaque. The extracellular portions of these molecules make up portions of the anchoring filament that probably contribute to the structure known as the subbasal dense plate, which underlies hemidesmosomes in the lamina lucida region. β4 integrin only pairs with the α6 subunit, whereas the α6 subunit can combine either with the β4 integrin or with the β1 integrin. Both the α6β1 or α6β4 integrin combinations have been shown to act as receptors for laminins, but only α6β4 integrin acts as a specific receptor for laminin-332. α6β4 integrin plays a central role in organization of the hemidesmosome. The β4 integrin contains an especially large cytoplasmic domain, which functions in the interaction with other proteins of the hemidesmosomal plaque, including collagen XVII and plectin.29 Skin from transgenic mice lacking β4 integrin is devoid of hemidesmosomes and shows severe deficits in cell adhesion.30 Interactions between plectin and α6β4 integrin appear to be critical both in the assembly as well as the disassembly of hemidesmosomes.31
Collagen XVII (BPAG2, BP180) is a collagenous protein with a type II transmembrane orientation. Based on electron microscopy and cross-linking studies, collagen XVII assembles into a triple-helical homotrimer and contains three main regions: (1) an intracellular amino-terminal globular head, (2) a central rod, and (3) an extracellular flexible tail.32 Collagen XVII associates with laminin-332 and α6β4 integrin in adhesion structures termed stable anchoring contacts. These stable anchoring contacts are formed by keratinocytes in vivo, and are thought to represent prehemidesmosomes.33 The autoantigen in linear immunoglobulin (Ig) A bullous dermatosis, LAD-134,35 is a 120-kD protein which has been shown by peptide sequencing to be the cleaved exodomain of collagen XVII.36 Collagen XVII undergoes processing in keratinocyte cultures and in skin through the action of sheddases, membrane-associated proteases that solubilize cell surface receptors.37–39
In addition to α6β4 integrin and collagen XVII, anchoring filaments contain the molecules laminin-332 and laminin-311. Like all members of the family of laminin proteins,40–42 laminin-332 is a large heterotrimeric molecule, and contains α3, β3, and γ2 chains.43,44 The first laminins to be described contained three short arms and one long arm, forming a cross shape as shown by rotary shadowing analysis. In contrast, laminin-332 contains truncations of each short arm.45–47 Because of these short arm truncations, laminin-332 cannot self-polymerize with other laminins or bind to nidogen. Instead, laminin-332 forms a disulfide-bonded attachment to laminin-311,48 the other known anchoring filament laminin49 which contains α3, β1, and γ1 chains. Laminin-332 also undergoes processing of its γ2 and α3 chains.50 While the rat laminin γ2 chain has been previously been shown to be processed by metalloproteinase-251 and membrane type metalloproteinase type 1,52 the predominant site of cleavage by these enzymes is not conserved in human laminin-332.53 Other studies have shown that processing of laminin γ2 chain takes place through a special class of proteins termed C-proteinases which also process C-terminal domains of procollagen molecules.54 While one member of this class of proteins, bone morphogenic protein 1,55 is capable of performing this action, a splice variant of bone morphogenic protein 1 (mammalian tolloid) is predominantly expressed in keratinocytes and fibroblasts and thus likely performs this function in the skin.53 Mammalian tolloid also processes the laminin α3 chain,53 although other enzymes such as plasmin,56 matrix metalloproteinase 251 or membrane type matrix metalloproteinase 152 are also capable of this function. The γ2 chain short arm appears important in the assembly of laminin-332 into basement membrane.57 The antigen recognized by mAb 19-DEJ-158 also localizes to anchoring filaments but its molecular identity remains unknown.
Collagen VII is the major constituent of anchoring fibrils. Analysis of the deduced amino acid sequence of collagen VII59 reveals the presence of a long central collagenous region characterized by repeating GlyXY sequences that contains a number of noncollagenous interruptions, including a 39 amino acid noncollagenous segment in the center of the helix which corresponds to the “hinge region” predicted by biochemical studies.60,61 These interruptions account for the flexibility of the collagen VII molecule, and explain its ability to loop around and entrap dermal matrix molecules, thus stabilizing the basement membrane to the underlying papillary dermis.62 A 50 KDa component of anchoring fibrils has also been identified which appears to localize to the insertion sites of anchoring fibrils to the lamina densa.63
The 145-kDa N-terminal end of collagen VII contains the largest noncollagenous domain, which inserts onto the lamina densa and anchoring plaques. Collagen IV, the most abundant component of these structures, binds to the collagen VII NC-1 domain. A direct interaction between anchoring filaments and anchoring fibrils exists from a specific interaction between the anchoring filament component laminin-332 and the collagen VII NC-1 domain.64,65 Collagen VII binds the β3 chain on laminin-332.64,66–68 This appears to be a critical factor in the maintenance of dermal–epidermal cohesion. Like all collagens, collagen VII assembles into a triple helix. Only one gene and one chain of collagen VII, the α1 chain, have been identified, and thus collagen VII is a homotrimer. Collagen VII triple helices are joined together at their processed NC2 globular domains to form antiparallel dimers.62,69 Processing of the NC-2 domains takes place via the same family of C-proteinases (bone morphogenic protein 1 and/or mammalian tolloid) that are known to process laminin-332, a closely associated molecule. Anchoring fibrils may derive from lateral associations of collagen VII antiparallel dimers.
Clinical Findings
Epidermolysis bullosa has been traditionally classified according to the level of BMZ separation on transmission electron microscopy into simplex, junctional and dystrophic subtypes,70 (Fig. 62-2). Although this ultrastructural “gold standard” for diagnostic grouping of EB has been electron microscopy, immunomapping of basement membrane antigens as viewed by indirect immunofluorescence can also be quite useful in distinguishing subtypes of EB as well as from other disorders in the differential (see Box 62-1) and is considerably less labor intensive. Within each of these groups there are several distinct types of EB based on clinical, genetic, histologic, and biochemical evaluation.71 These findings are summarized below (Table 62-1).
Figure 62-2
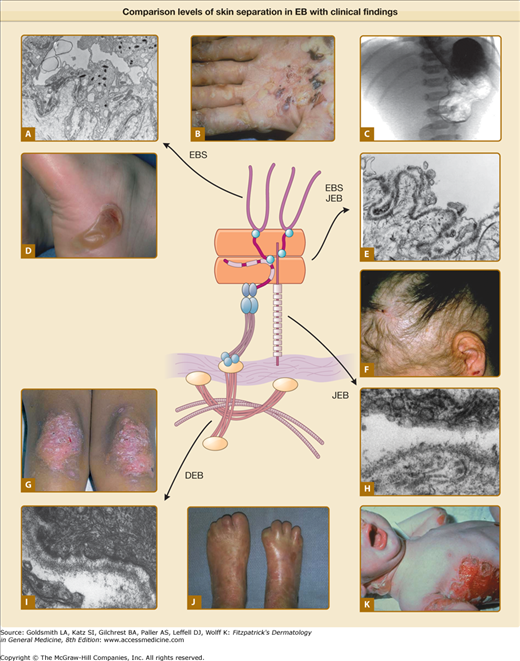
Comparison of levels of skin separation in EB with clinical findings. A. Transmission electron micrograph showing typical intraepidermal separation in EBS. B. Palmar hyperkeratosis and erosions in EB herpetiformis. C. Radiograph showing pyloric atresia associated with EB. D. Localized blister on heel in EBS Weber–Cockayne. E. Transmission electron micrograph showing typical separation at level of hemidesmosome in EB with pyloric atresia, alternatively classified as EBS or JEB. F. Nonscarring diffuse alopecia and scalp erosions in generalized atrophic benign epidermolysis bullosa. G. Localized dystrophic changes with milia in dominant DEB. H. Transmission electron microscopy showing typical intralamina lucida separation in JEB. I. Transmission electron microscopy showing typical sublamina densa separation in DEB. J. Pseudosyndactly in recessive DEB. K. Generalized blistering in Herlitz JEB. EBS = epidermolysis bullosa simplex; DEB = dystrophic epidermolysis bullosa; JEB = junctional epidermolysis bullosa.
Most Likely
|
Consider
|
Always Rule Out
|
Level of Separation | Disease | Defect |
|---|---|---|
Simplex | Generalized | KRT5/KRT14 |
Simplex | Dowling–Meara | KRT5/KRT14 |
Simplex | Localized | KRT5/KRT14 |
Simplex | Ogna | KRT5/KRT14/PLEC1 |
Simplex | Mottled pigmentation | KRT5/KRT14 |
Simplex | EB with muscular dystrophy | PLEC1 |
Simplex | Superficialis | KRT5/KRT14 |
Simplex | Ectodermal dysplasia-skin fragility | PKP1 |
Junctionala | EB with pyloric atresias | ITGB4/ITGA6/PLEC1 |
Junctional | Herlitz | LAMB3/LAMA3/LAMΓ2 |
Junctional | Non-Herlitz | LAMB3/LAMA3/LAMG2/COL17A1 |
Junctional | Localized | COL17A1 |
Dystrophic | Generalized dominant | COL7A1 |
Dystrophic | Localized dominant | COL7A1 |
Dystrophic | Recessive | COL7A1 |
Variable | Kindler syndrome | KIND1 |
Epidermolysis bullosa simplex (EBS) is a disease group characterized by intraepidermal blistering and most often is associated with keratin gene mutations. The disease phenotypes range from mild to severe among different subgroups.72 The common EBS types are dominantly inherited and include Dowling–Meara (Herpetiformis) EBS, generalized (Koebner) EBS, and localized (Weber–Cockayne) EBS. There are several uncommon varieties that include EBS Ogna, EBS with muscular dystrophy, EBS with mottled pigmentation, EBS associated with BP230/BPAG1 mutation, and a group of suprabasal EBS subtypes. EBS is not usually associated with growth retardation or anemia.
This subtype presents at birth, has a generalized distribution and is regarded as the most severe of the EBS subtypes (Fig. 62-3). However, it differs from the Koebner variant in that the oral mucosa is more often involved, occasionally showing extensive erosions. Milium formation may sometimes occur in infancy in patients with this subtype; however, this postwound phenomenon usually resolves after infancy. The disease can often be associated with spontaneous appearance of grouped or “herpetiform” blisters. These occur on the trunk and proximal extremities and heal without scarring. It should be noted that this herpetiform pattern may not be seen when patients show a generalized blistering. Therefore, its absence should not be used as a basis to exclude this EB subtype. Dowling–Meara EBS often shows nail involvement, with shedding and regrowth with dystrophy or long nails with subungual hyperkeratosis. Hyperkeratosis of the palms and soles often develops beginning in early childhood and can progress to confluent keratoderma of the palms and soles. These can be quite painful and, occasionally, interference with ambulation has led to flexural contractures. Esophageal involvement in Dowling–Meara has occasionally been reported, and ranges ranging from erosions to pyloric atresia.73 The upper respiratory tract can also be affected, including the laryngeal mucosa.74 Natal teeth have been described.
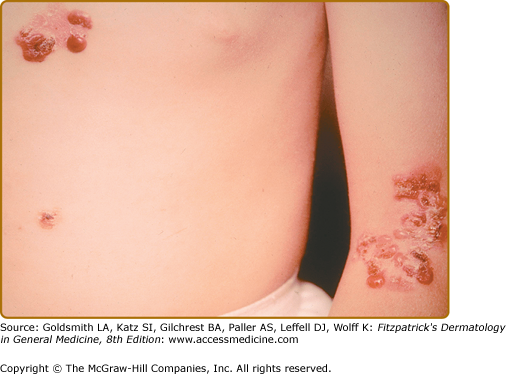
The other common form of generalized EBS is also known as the Koebner EBS. This subtype shows an onset of generalized blistering at birth or at latest during early infancy. The hands, feet, and extremities usually show the most involvement. Lesions often heal with postinflammatory hyper- or hypopigmentation. Atrophy and milia can occur, but are much less frequent than in Dowling–Meara EBS. Palmoplantar hyperkeratosis and erosions may be present (Fig. 62-4). Thickening of the soles is common, but often does not present until later childhood. The oral mucosa sometimes shows mild erosive activity but these usually improve with increasing age.
This is the mildest form of EB and is often referred to as the Weber–Cockayne subtype of EBS (Fig. 62-5). This disease is the most common form of EB and often presents during infancy or childhood. Occasionally, it presents in early adulthood, such as when blisters are noted following marching during military service. It is speculated that there are a number of undiagnosed cases of this form of EB, as it can be mild enough to escape reporting or detection during clinical visits. Hyperhidrosis of the palms and soles is a common association. Blisters can occasionally become secondarily infected. Postinflammatory pigmentary abnormalities occur with this variant, but milia and scarring as a rule are absent. Blistering activity usually follows areas of trauma, with hands and feet being the most common and the scalp being the least common. Mild oral erosions are present only rarely and usually resolve with increasing age. Nail involvement is rare with this EB subtype.
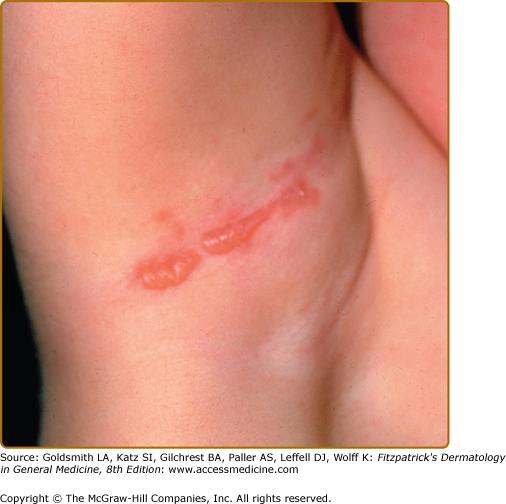
Onset in infancy is common with seasonal blistering (summer) on the acral areas. Small hemorrhagic and serous blisters occur primarily on the extremities. Healing occurs without scarring. This disease was originally reported in patients from Norway. These patients also show a characteristic onychogryphosis of the great toenails.
This rare clinical entity is an autosomal recessive disorder which consists of generalized blistering of the skin at birth or shortly thereafter. This is accompanied by a progressive muscular dystrophy.75 It presents with generalized blistering similar to EBS of Koebner clinically. These patients have been shown to harbor mutations in the gene coding for HD1/plectin.76–78 Mutations in plectin can also cause EB with pyloric atresia (see Junctional EB with Pyloric Atresia) and be difficult to distinguish clinically from disease resulting from integrin mutations.
This form of EB, as its name implies, is characterized by mottled hyperpigmentation of the trunk and proximal extremities. Blistering tends to be mild, and begins at birth or early infancy. Pigmentary alterations appear reticulated and increase with advancing age, as blistering may improve. Mild oral mucosal involvement may be present in infancy. This progressive pigmentation is distinct from the large melanocytic nevi, which can be seen in all three EB types.79
EBS superficialis is an uncommon form of EBS, named after the subcorneal separation that produces the blisters in this disease.80 Erosions and crusts, rather than intact bullae, are usually seen in these patients, and heal with postinflammatory pigmentary changes. Despite the superficial cleavage plane, nail involvement. Atrophic scarring and milia have been observed in this disease.
Lethal acantholytic EBS81 is a rare recessively inherited and lethal disorder characterized by generalized erosions at birth. As in the superficialis subtype, intact blisters are not normally seen, due to the very superficial level of epidermal separation, which appears sheet-like. Nails are dystrophic to absent. Complete alopecia, neonatal teeth, oral erosions, and respiratory involvement distinguish this disorder from other superficial EBS subtypes.
Ectodermal dysplasia skin fragility syndrome is another inherited disorder of suprabasilar epidermal separation, characterized by generalized erosions and sometimes superficial blisters at birth.82 Alopecia is characteristic, as are palmoplantar keratoderma, painful fissures and nail dystrophy. Patients may sometimes demonstrate failure to thrive, cheilitis, hypohidrosis, and pruritus as well. Like the other superficial EBS subtypes, this disorder is associated with dystrophic nails.
Most of the patients with EB simplex analyzed at the genetic level have been found to be associated with mutations of the genes coding for keratins 5 and 14.20,72 The level of separation of the skin in these patients is at the midbasal cell, shown in Fig. 62-2A, associated with variable intermediate filament clumping. Hemidesmosomes and other BMZ structures are normal by electron microscopy. The majority of keratin gene mutations associated with epidermolysis bullosa simplex are dominantly inherited due to abnormalities in the multimeric assembly of keratin filaments. There is a smaller subset of patients with recessively inherited disease of varying severity.83,84
Mutations coding for the most highly conserved regions of keratins 5 and 14, the helix boundary domains,85 correlate with the most severe form of EBS, the Dowling–Meara subtype, which exhibits intermediate filament clumping seen by transmission electron microscopy. On the other hand, milder types of disease, such as the Weber–Cockayne subtype, are associated with mutations coding for regions of keratins 5 and 14 that are less conserved. Mutations that code for a specific region of the amino-terminus of keratin 5 are present in patients having EBS with mottled pigmentation.86 Although significance of this type of mutation and its association with pigmentary abnormalities remains unclear,87,88 it has been suggested that the keratin 5 globular head domain is responsible for keratin filament insertion onto melanosomes. Recent studies have shown that some mutations of the keratin 5 gene may produce protein which is unstable under increased temperatures.89 This could help to explain the well-observed exacerbation of some subtypes of EBS to warm temperatures.
The mutations associated with EBS with muscular dystrophy produce premature termination codons, splice site, or other mutations which result in lack of expression or defective expression of plectin.90 Although the form of EB associated with plectin abnormalities is classified as simplex, it has an identical level of skin separation to that seen in junctional epidermolysis bullosa (JEB) with pyloric atresia. Specifically the separation is present just above the level of the hemidesmosome in the intracellular part of the BMZ. This separation of EBS with muscular dystrophy and JEB with pyloric atresia, diseases with identical levels of separation, into two distinct EB categories illustrates the limitations of the current EB classification system. Plectin defects, like α6β4 integrin defects, can also be associated with pyloric atresia.90
Plectin is normally expressed in a wide range of tissues, including muscle.21 While the mechanism of muscular dystrophy in plectin-deficient patients is unknown, it has been observed that disorganization of muscle sarcomeres occurs in the absence of plectin. It is possible that absence of plectin’s spectrin-like domain, which may normally interact with actin filaments in muscle, may be a key factor in the muscle pathology.91 Interestingly, a case report of autosomal recessive EBS has been recently identified to be associated with both muscular dystrophy and pyloric atresia.92 This case was associated with homozygous premature termination codon mutations in the plectin gene causing a significant loss of plectin expression in the skin.92
Recently, another unique case of recessive dominant EBS has been identified, this one associated with null mutations of the dystonin gene coding for BP230/BPAG1.93 Inherited blistering/skin fragility described with this defect was associated ultrastructurally by loss of hemidesmosomal inner dense plate and a concomitant reduction of other hemidesmosomal proteins, presumably by loss of BP230 stabilization. This patient’s blistering was also accompanied by a neuropathy, which may be part of the inherited defect, since a splice variant of this molecule has a neural distribution.
The underlying molecular defect of ectodermal dysplasia skin fragility syndrome has been shown to be loss of function of the desmosomal protein plakophilin 1, encoded by PKP1.94,95–100 Plakophilin is expressed mainly in suprabasilar keratinocytes and outer root sheath cells. Microscopic findings in this disease are usually intraepidermal acantholysis, located in the areas where plakophilin 1 is normally expressed.
All patients with JEB share the common histopathologic feature of blister formation within the lamina lucida of the BMZ due to defects of anchoring filaments located in the lamina lucida and superior lamina densa. This group of diseases is inherited in an autosomal recessive manner and there is considerable variation of the individual clinical phenotypes depending on the molecular defect. Three principal forms of JEB are most common. Herlitz disease, JEB gravis or lethal JEB are each used to describe patients presenting with the most severe and most common phenotype.14,101
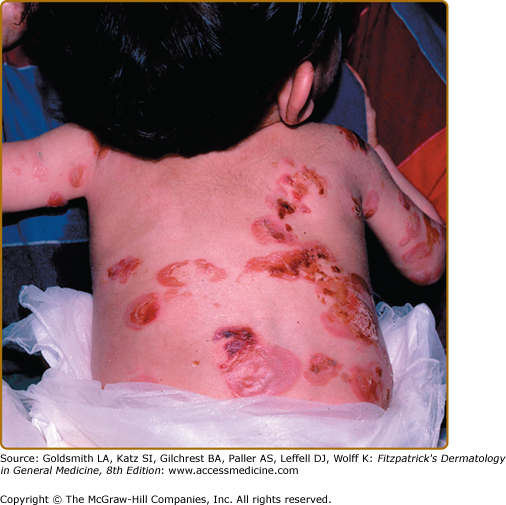
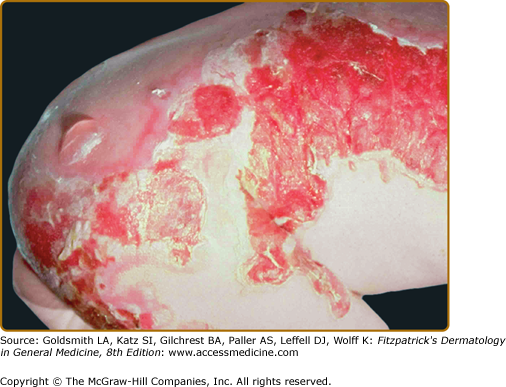
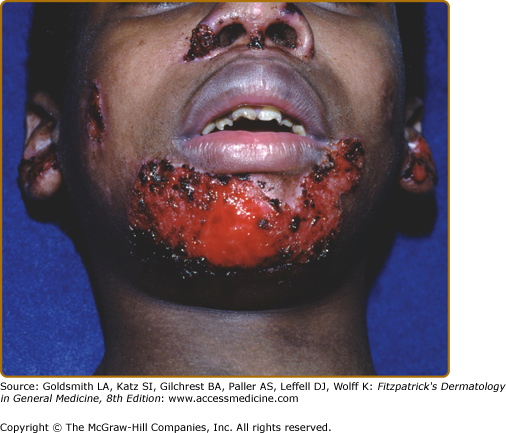
Patients with Herlitz JEB, also known as JEB letalis, is one of the most severe EB subtypes and is lethal in most affected children during infancy or early childhood.102 This disorder is characterized by generalized and often extensive blistering at birth (Fig. 62-6). Later during infancy, a distinctive periorificial granulation tissue begins to manifest, primarily around the mouth, eyes, and nares. The scalp, periauricular areas and, less frequently, locations outside of the head and neck may also be affected. This hypertrophic granulation tissue much less commonly occurs in nonlethal subtypes. Nails are usually severely affected and often are lost during infancy. The presence of nail involvement with periungual hypertrophic granulation tissue during the neonatal period can be a clue to this diagnosis. When nails are still present, they are usually dystrophic. Tooth enamel pitting is characteristic in both primary and secondary teeth, and can progress after tooth eruption. Oropharyngeal mucosal erosions are usually present and may be widespread. Erosions of all stratified squamous epithelial tissues, including nasal, conjunctival, esophageal, tracheal, laryngeal, rectal, and urethral mucosa can be affected. Associated systemic findings in severe cases are important factors in the lethality of this disease. Involvement of the large airways, including tracheolaryngeal stenosis or obstruction, is commonly associated with Herlitz JEB disease and hoarseness in early infancy is an ominous sign. There is a characteristic failure to thrive and growth retardation in this disease, often with a mixed anemia. Sepsis is a common and often lethal complication.
(Figs. 62-7 and 62-8).
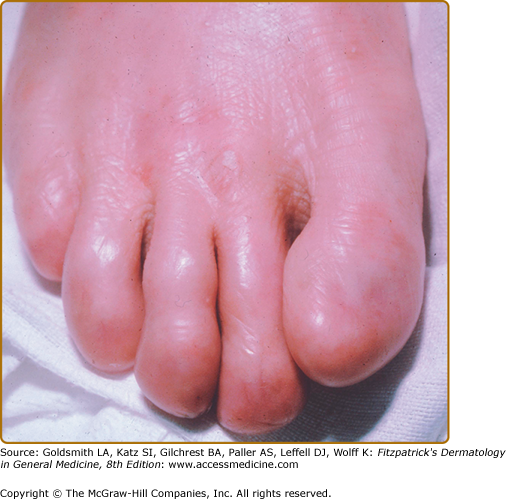
Sometimes patients initially presenting with a Herlitz-like phenotype will survive infancy and clinically improve with age. These patients eventually prove to have a severity of blistering and oral erosions less than in the lethal form. In particular, a lack of significant hoarseness is regarded as a favorable prognostic sign, indicative of less severe internal disease manifestations. Scalp and nail lesions, as well as periorificial nonhealing erosions with exuberant granulation tissue are among the most common findings in these patients during childhood. Despite the lack of lethality in infancy, these patients nonetheless can have severe epithelial adhesion abnormalities. Tracheostomies or gastrostomy tubes may help in patient survival. The nonlethal status of these patients distinguishes them from the Herlitz group and the terms nonlethal JEB or JEB mitis have been used in the past to describe these patients. They are much less common than Herlitz EB patients. There are other rare variants of nonlethal JEB that present with localized junctional blistering of the extremities or intertriginous areas.
Interestingly, the association of hypertrophic granulation tissue and laminin-332 defects has been noted in another group of patients with mutations of the laminin α3 (LAMA3) gene leading to a small truncation of the N-terminal IIIa domain of the laminin α3 chain. This disease, termed laryngo-onycho-cutaneous syndrome, is characterized by a triad of cutaneous erosions, nail dystrophy, and exuberant mucocutaneous granulation tissue, especially in the conjunctiva and larynx.103 An experimental animal model of engineered laminin-332 deletions has been recently described which recapitulates this hypergranulation tissue phenotype.104
Generalized atrophic benign EB (GABEB) is a distinct subset of non-Herlitz JEB that also presents at birth with generalized cutaneous involvement.105 Despite the widespread cutaneous blistering, there is a relative paucity of oral erosions or other mucosal disease. While enamel pitting is present, resulting in extensive dental caries, and nail dystrophy can often be severe, there is little other extracutaenous involvement noted in these patients. The blisters in these patients, which heal with a characteristic atrophic scarring, are debilitating and can be widespread, but nonetheless these patients generally have a normal lifespan. Blistering improves with age, growth is normal and anemia is uncommon. Some patients with this disease have undergone normal uncomplicated pregnancies and deliveries. One peculiar characteristic of these patients is a progressive alopecia of the scalp and terminal hairs elsewhere in the body. The hair loss starts to become severe after the onset of puberty. While patchy hair loss associated with atrophic scarring has been described, often the alopecia is quite diffuse and scarring is subtle or nonexistent.
A very rare subtype of JEB is the localized variant, also known as minimus JEB. These patients generally show mild disease that can be accentuated in localized areas, most often the hands, feet and pretibial regions. Nails can sometimes be shed or become dystrophic, and enamel pitting can occur. Oral or nasal erosions can also occur; however, there is absence of any internal involvement. These patients generally have a favorable prognosis and a normal lifespan.
These patients exhibit extreme mucosal and cutaneous fragility and may also have various urological abnormalities, including hydronephrosis, and nephritis, and rudimentary ears. Mutations of the genes coding for the β4 and α6 integrin are associated with EB106 and in which pyloric atresia. Plectin mutations can also be associated with EB and pyloric atresia, although the the integrin mutations are more reliably associated with pyloric atresia. In these patients, hemidesmosomes are usually absent or rudimentary, and the level of separation is intraepidermal at the level of the hemidesmosome as seen in EBS with muscular dystrophy described above. The skin ultrastructure in patients with α6β4 defects is remarkably similar to the β4 integrin knockout mice described above; however, the mice do not show pyloric atresia. These studies illustrate well that there are limitations in the utility of animal models to study human epidermolysis bullosa. Most cases of this disease are quite severe and lethal in infancy, due to extensive extracutaneous epithelial sloughing, in addition to widespread blistering of the skin and mucosa. Rare nonlethal cases of this disease have been characterized which appear to result from a partial loss of function of β4 integrin.107 Interestingly, nonlethal JEB can sometimes ameliorate itself through alterations in mRNA splicing.108,109
JEB can be associated with mutations of the genes coding for any of the α3, β3, or γ2 subunits of laminin-332.110,111 Absence of any of the three chains results in a lack of trimeric laminin-332 assembly and secretion, which results in a similar blistering phenotype. Patients with mutations of genes coding for the α3 or γ2 laminin subunits will still show normal expression of laminin-311, which contains α3, β1, and γ1 chains.44 Therefore, positive linear BMZ staining of JEB skin for the α3 chain on immunofluorescence mapping studies with absent expression of the other chains is an indication of either a α3 or γ2 chain defect. Conversely, absence of α3 staining on immunofluorescence can be indicative of a mutation of the α3 gene. About 80% of laminin-332 mutations can be traced to one of two recurrent nonsense mutations in the LAMB3 gene, making prenatal testing for laminin-332 lesions easier than other EB candidate genes.112 In the Herlitz patients, all of the mutations so far detected have been those producing premature termination codons, resulting in absence of expression of laminin-332. While Herlitz cases generally show a complete lack of expression of laminin-332, non-Herlitz patients with abnormal granulation tissue and significant mucosal involvement often show reduced expression of laminin-332. This is due to deletions or missense mutations that result in partial loss of laminin-332 function.113–115 In some cases, spontaneous amelioration of blistering in severe junctional epidermolysis bullosa cases has taken place, leading to the expression of laminin-332.109
In the GABEB variant of non-Herlitz JEB, blistering occurs in the lamina lucida region and abnormalities of hemidesmosomes/anchoring filaments are usually present (Fig. 62-3). While laminin-332 mutations underlie a subset of GABEB patients, the majority of these patients have abnormalities of the hemidesmosomal protein collagen XVII (also known as BP180 or BPAG2). A number of mutations of the gene coding for collagen XVII have been described in GABEB patients, including premature termination codon mutations, missense mutations, splice site mutations, truncations, and a glycine substitution mutation.111,116 While intralamina lucida/junctional skin separation has been shown in all patients with this disease, one patient was described with a cytoplasmic deletion of collagen XVII who showed intrabasal epidermal skin separation.117 Localized junctional EB has been shown to be associated with COL17A1 mutations.118
Of interest, mosaic GABEB patients have been identified who demonstrate well-defined areas of blistering associated with absence of collagen XVII expression as well as areas of nonblistering skin associated with normal collagen XVII expression. Careful analysis of these patients’ keratinocytes revealed reversion of one of the two alleles of the mutation, most likely due to a mitotic gene conversion involving nonreciprocal exchange of parental allele DNA.119,120 It is possible that this phenomenon of mutational rescue of genes affecting recessive cases of EB is more widespread that currently reported and more careful examination of involved and uninvolved regions of EB skin undoubtedly will reveal more cases. Understanding how epidermolysis bullosa can undergo spontaneous molecular correction (revertant mosaicism) in these cases could help in the design of future molecular therapeutic strategies.
Dystrophic EB (DEB) is characterized by blisters that heal with scarring and milium formation. DEB can be inherited either in an autosomal recessive (RDEB) or dominant fashion (DDEB). One of the most important reasons to distinguish between these two subtypes is the increased prevalence of invasive squamous carcinoma associated primarily with the recessive form. Regardless of the mode of inheritance, DEB is derived from defects of the ultrastructural entity known as the anchoring fibril, which results in sublamina densa separation.
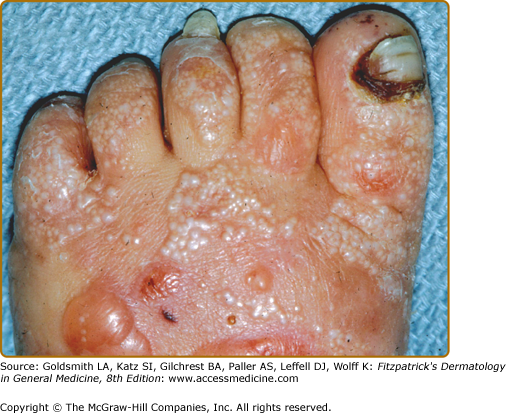
The localized subtype of dominant DDEB (sometimes called the Cockayne–Touraine of DDEB) can present at birth, but occasionally it is not appreciated until childhood. While generalized blistering can take place especially early in life, the blistering usually becomes localized to repetitively traumatized areas such as knees, sacrum, and acral surfaces (Fig. 62-9). These areas show a characteristic scarred, dystrophic appearance. Often the scarring is hypertrophic. Milia are common accompanying features of the healing process in these patients. Nail dystrophy or nail loss with atrophic scarring of the distal digits are common. Occasionally, nail abnormalities can be the only presenting abnormality in DDEB. Oral lesions are not common and teeth are usually unaffected. These patients have a good prognosis and a normal lifespan.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


