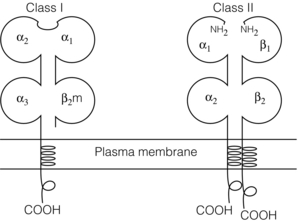
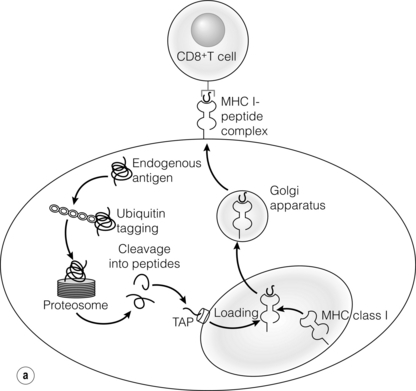
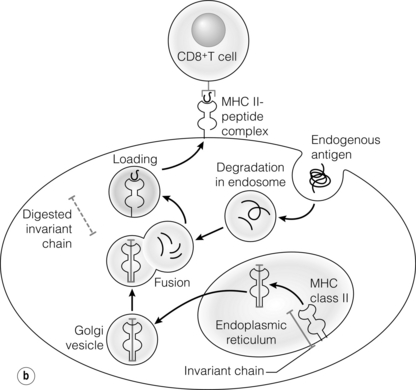
3 In Matzinger’s model of immune activation, ‘danger’ to the host (danger-associated molecular patterns, DAMP) is sensed through pattern recognition receptors (PRRs) on antigen-presenting cells (APCs).1,2 APCs are made up of a variety of innate cells, including tissue resident macrophages and dendritic cells (DCs). APCs engulf, process and present peptide antigen on their cell surface in the context of major histocompatibility complex (MHC) antigens. They then migrate to lymphoid tissue and present the antigen to T cells. The resulting adaptive response is determined by the balance between co-stimulatory and co-inhibitory signals received alongside this specific antigenic stimulus; this will be discussed in more depth later. Histocompatibility antigens were first defined on the basis of their preventing transplantation between outbred members of the same species. In pioneering work, Snell and Higgins identified that the genes responsible for rejection were found on chromosome 17 at just one locus (H2); this complex of murine genes was termed the major histocompatibility complex (MHC).3 The characterisation of MHC in humans was later identified at the same locus and the nomenclature of human leucocyte antigens (HLAs) was adopted after the first Histocompatibility meeting in 1964. Differences between donor and recipient MHC antigens significantly contribute to rejection, despite significant progress in immunosuppressive therapy.4 Furthermore, numerous minor histocompatibility systems also exist and can result in allograft rejection; this is particularly problematic in bone marrow transplantation (see Chapter 4). There are two main types of MHC molecules, MHC class I and II (see Fig. 3.1). In humans, the genetic sequences for these glycoproteins are found on the short arm of chromosome 6, with the exception of the light chain β2-microglobulin, which is encoded on chromosome 15. There are six main loci in the human MHC: HLA-A, HLA-B and HLA-C belong to HLA class I, while HLA-DP, HLA-DQ and HLA-DR belong to HLA class II. Figure 3.1 Structure of MHC class I and II molecules. α1, α2, α3 of MHC class I are membrane integral and form a non-covalent bond with β2-microglobulin 9 (β2-M), which is not membrane bound. MHC class II is formed from α and β chains, each with two domains and each membrane integral. MHC class I proteins are cell-surface glycoproteins expressed on all nucleated cells and present peptides derived from intracellular proteins. They are formed by a highly polymorphic heavy chain (45 kDa) and a less variable light chain, β2-microglobulin, which is bound non-covalently to the heavy chain. Endogenous antigens are digested into peptides by proteosomes, and loaded on to class I MHC, as shown in Fig. 3.2a. MHC class I proteins present antigen to CD8-positive (cyotoxic) T cells, which have important roles in the defence against viral infection and malignancy. Figure 3.2 Antigen processing and presentation in the MHC class I and II pathways. (a) Processing of endogenous antigens occurs primarily via the class I pathway. Peptides are produced and loaded into MHC class I proteins as shown. (b) Processing of exogenous antigens occurs primarily via the class II pathway. Antigens are taken up into intracellular vesicles. MHC class II proteins are synthesised in the endoplasmic reticulum, and fuse with vesicles containing peptide. MHC, major histocompatibility complex; TAP, transporters associated with antigen processing. MHC class II proteins are found constitutively on the surface of APCs and B cells, but can also be induced by the process of inflammation on endothelial cells. Exogenous antigen is engulfed, degraded within endosomes and presented on the cell surface in the context of MHC class II (see Fig. 3.2b). Antigen processed in this way is recognised by CD4-positive (helper) T cells; these cells are important in the defence against extracellular infection and have been linked to the development of autoimmunity. The specific structure of the binding groove of MHC class I or II determines which peptide can be bound to it. In evolutionary terms, the extreme polymorphism in MHC proteins probably reflects the need to bind a vast array of rapidly mutating pathogens. The assembly of the MHC–peptide complex, as shown in Fig. 3.2, involves a sophisticated mechanism of antigen uptake, processing and transport. MHC class I chain-related A (MICA) antigens are surface glycoproteins with functions related to innate immunity.5 The genes are located within the class I region of the chromosome and are highly polymorphic. The contribution of MICA antigens to rejection is controversial, with some studies showing no effect on rejection,6 while other large studies have demonstrated that the presence of MICA antibodies was associated with rejection.7,8 Minor histocompatibility antigens (miH) are distinct from MHCs; they are less polymorphic and typically induce a weaker response than disparities in the MHC. The male-specific H-Y antigen is the most well-defined miH antigen and has been shown to play a role in the rejection of bone marrow and in rodent models of transplantation. The role in solid organ transplantation is not yet fully understood, with some studies suggesting that it may only have an effect on short- but not long-term outcomes following renal transplantation.9 Inflammation lies at the centre of rejection and begins even prior to organ transplantation, with the haemodynamic and neuroendocrine responses associated with brain stem death, the process of multiorgan retrieval and subsequent cold preservation (see Fig. 3.3). During the transplantation procedure itself, there is a further period of warm ischaemia and reperfusion. This compound injury results in the release of DAMPs and stimulates an inflammatory response, which promotes and shapes alloantigen-specific immunity and has an effect on long-term graft survival.10 The importance of these events in transplantation is illustrated by the superior outcome of live donor transplants even in the face of significant MHC mismatch, the impact of cold ischaemia time on graft outcome11 and the higher rates of rejection observed in individuals with delayed graft function.12 Indeed, in experimental syngeneic transplantation, graft histology very similar to that seen in chronic allograft damage may be reproduced by prolonged ischaemia. Ischaemia–reperfusion injury (IRI) is an inevitable consequence of organ retrieval as the organ is temporarily deprived of its blood supply before being reperfused and reoxygenated following transplantation. IRI is responsible for a spectrum of early organ dysfunction after transplantation and the mechanisms involved are summarised in Fig. 3.4. Figure 3.4 Mechanisms of ischaemia reperfusion injury. (1) Anaerobic metabolism. (2) Breakdown of Na+/K+ pump, with resultant cellular oedema. (3) Acidosis leads to Ca2 + influx, and mitochondrial injury. (4) Activation of phospholipase A2 and caspases. (5) Increased MHC class II expression, which intensifies the immune response. Cessation of arterial supply results in oxygen deprivation and a switch to anaerobic metabolic pathways. There is also a build-up of toxic metabolic byproducts, including lactate, H+ and inorganic phosphate. High-energy phosphates, such as adenosine triphosphate (ATP), are vital for most cellular functions. Their depletion inhibits the Na+/K+ pump, thereby impairing the ability of the cell to maintain its membrane potential and subsequently the integrity of the cell membrane.13 Acidosis occurs as an immediate result of anaerobic glycolysis and has been shown to correlate well with tissue injury.14 This is one of a number of mechanisms that causes an influx of calcium into the cell, leading to mitochondrial injury as well as activation of proteases and phospholipases, including caspases and phospholipase A2. The generation of free radicals also contributes directly to tissue injury. Ischaemia induces an acute inflammatory response, leading to endothelial activation with increased permeability and adhesion molecule expression. On reperfusion, circulating inflammatory cells become adherent to the activated epithelium, with a resulting increase in release and expression of proinflammatory mediators (cytokines, chemokines and adhesion molecules), leading to further recruitment and activation of leucocytes.15,16 During the hours or days following the ischaemic insult, repair and regeneration occur alongside apoptosis (characterised by nuclear fragmentation, plasma membrane blebbing and cell shrinkage), autophagy cell-associated death (with loss of organelles and accumulation of vacuoles) and necrosis (with plasma membrane rupture and leakage of proteases extracellularly). The fate of the organ will depend on whether regeneration or cell death prevails.17 IRI is a form of sterile inflammation, in which endogenous DAMPs are released into the extracellular compartment as a result of tissue injury.18 These DAMPs signal through various pattern-recognition receptors, such as Toll-like receptors (TLRs), and result in the activation of effector pathways, including the production of proinflammatory cytokines and chemokines.19 As a result of natural antibodies, innate recognition proteins can react to self in a process known as innate autoimmunity; this leads to the activation of complement. The complement system is an immune surveillance system set up to detect cellular debris, apoptotic cells and non-self, and plays an important role in the amplification of the immune response seen in sterile inflammation, and as such is an attractive target for therapy.20,21 There is evidence for the role of CD4+ T cells in the pathogenesis of IRI. This has been shown in a number of models of IRI, including renal,22 hepatic,23 cardiac24 and cerebral.25 In contrast, regulatory T cells may have a protective effect in IRI, with evidence emerging in models of ischaemic stroke26 and renal IRI.27 Through this wide range of mechanisms, IRI leads to increased expression of HLA antigens by the graft, as well as the release of proinflammatory chemokines, cytokines and adhesion molecules within the graft; this leads to increased recruitment of effector immune cells into the graft and is likely to increase the risk of rejection.10 Recognition of alloantigen by T cells Alloimmunity refers to the initiation of an immune response against antigen from the same species. The most common form of alloimmune injury is initiated when donor alloantigens are presented to recipient T cells by APCs. T cells engage MHC with their receptors either directly on donor-derived APCs, or indirectly as processed peptide in the context of self-MHC. A third ‘semi-direct’ pathway has more recently been elucidated, and all three are illustrated in Fig. 3.5. Donor-derived APCs (typically dendritic cells), known as passenger leucocytes, carry donor antigens from the transplanted organ to the recipient’s draining lymph nodes. These donor-derived APCs express high levels of MHC class I and II laden with antigen and are capable of activating both CD4+ and CD8+ T cells. This pathway of allorecognition is characterised by a high frequency of responder T cells (100-fold greater than for a response to conventional antigens) and is critical in the initiation of the alloresponse. CD4+ T cells with exclusively direct allospecificity can effect transplant rejection, in the absence of the indirect pathway, and CD8+ cytotoxic T cells,28 and indeed may contribute to the development of chronic rejection.29 The indirect pathway refers to recognition of processed peptides of allogeneic histocompatibility antigens presented by self-MHC in a restricted manner.30 Thus, donor protein is processed and presented in the context of MHC class II by recipient APCs, such as dendritic cells and macrophages, to CD4+ T cells. There is evidence to demonstrate the involvement of the indirect pathway in both acute31,32 and chronic33 graft rejection. These two seemingly distinct pathways of allorecognition suggest that there is no crosstalk between T cells activated by direct or indirect means. However, CD4+ T cells with indirect anti-donor specificity can amplify or reduce direct pathway CD8+ T-cell responses.34 This observation points to interactions between direct and indirectly activated T cells during graft rejection. Thus, a third semi-direct pathway has been proposed, in which recipient APCs can acquire functional MHC molecules from donor cells and this acquired MHC is capable of stimulating antigen-specific T-cell responses.35 This allows a single APC to stimulate both direct CD8+ T cells and indirect pathway CD4+ T cells.36 Recent work supports this hypothesis, demonstrating the presence of cells expressing both donor MHC class I and II molecules, allowing indirectly activated CD4+ T cells to regulate directly activated CD8+ T cells.37 T cells require two signals to stimulate T-cell activation: signal 1 is the antigen-specific binding of the MHC–peptide complex to the T-cell receptor (TCR) and signal 2 is the engagement of the co-stimulatory molecule. Signalling through the TCR alone is insufficient in itself to lead to T-cell activation and in the absence of co-stimulation (signal 2) may lead to anergy38,39 (a state of immune unresponsiveness). Targeting co-stimulatory pathways is clearly attractive for anti-rejection therapy and has been the focus of considerable interest both experimentally and in clinical trials (see Chapter 5). CD28 is the best characterised co-stimulatory molecule and is critical for T-cell proliferation and survival. CD28 is expressed on the surface of T cells and has two main ligands: CD80 (B7-1), which is constitutively expressed, and CD86 (B7-2), which is rapidly up-regulated on activated APCs.40 CD28 co-signals can stabilise messenger RNA of cytokines and amplify the activation of nuclear factor of activated T cells (NFAT) and nuclear factor κB (NFκB).41 More recently, another T-cell surface receptor that binds the same ligands as CD28 has been identified, known as cytotoxic T-lymphocyte antigen-4 (CTLA-4).42 This is up-regulated on T-cell activation and binds to B7-1 and B7-2 with much higher avidity than CD28. In contrast to CD28, CTLA-4 dampens T-cell responses by several mechanisms: it reverses the ‘stop signal’ needed for firm contact between T cells and APC, thus limiting their contact,43 and blocks the formation of microclusters of kinases that is required for effective transmission of signals from the TCR complex.44 In addition, there is evidence to suggest that CTLA-4 is required for optimal function of regulatory T cells (Tregs).45 Other co-inhibitory pathways exist and another potential therapeutic target is a second member of the CD28–CTLA-4 family, programme death-1 receptor (PD-1). Signalling through PD-1 leads to negative regulation of T- and B-cell activity, with decreased proliferation and cytokine production.46,47 In a murine model of hepatic IRI, blockade of signalling through PD-1 led to a worse injury.48 Such co-inhibitory pathways are attractive targets for therapy. The first strategy adopted was to develop a soluble recombinant fusion protein–CTLA–4Ig (abatacept), which was potent at inhibiting B7-1- but not B7-2-mediated T-cell responses.49 A modified version of CTLA–4Ig, belatacept, was subsequently developed through mutagenesis.50 In the seminal non-human primate study, Larsen and colleagues tested belatacept as monotherapy and in combination with either basiliximab or MMF and steroids. Belatacept was superior to CTLA-4Ig monotherapy in maintaining renal allograft survival. However, renal function declined during ongoing belatacept treatment, highlighting the need for adjunctive therapies. When used in combination with basiliximab induction, MMF and steroids, belatacept was shown to be safe and efficacious.50 Recent phase II and III clinical trials have yielded promising results, and this strategy holds promise to improve the care of transplant patients.51 • phospholipase C-γ1 activates NFAT; • protein kinase C-θ, which acts through NFκB and activator protein 1 (AP1); • renin–angiotensin system (RAS)–guanosine-releasing protein (GRP) and the growth factor receptor bound protein 2 complex, which act through RAS. These signalling pathways coordinate to drive proliferation and differentiation of the T lymphocytes, and can be influenced by different co-stimulatory molecules. In alloimmune responses, naive helper T cells are polarised towards various cell lineages, including Th1, Th2, Th17 and regulatory T cells (see below), defined according to the cytokine profile they produce (Fig. 3.6); this is referred to as signal 3. The antigen load, state of maturation of APCs, as well as the polarisation of T cells to a particular effector or regulatory lineage, ultimately determine the type of effector response and its effect on the transplanted organ. Th17 immunity is characterised by the production of IL-17, IL-21 and IL-22, with subsequent release of a host of proinflammatory cytokines (IL-6, IL-8, tumour necrosis factor (TNF)-α) and chemokines (such as CXCL1, CXCL2, CCL2 and CCL20).52 IL-17 is a potent stimulator and recruitor of innate immune cells, including neutrophils and monocytes. IL-17-producing cells have been detected in renal allografts undergoing rejection.53 There is evidence to suggest that they may play a role in acute rejection, particularly when other immune responses are suppressed by conventional immunosuppression.54 There is increasing recognition that Tregs are important in self-tolerance and may play a role in transplant tolerance. The manipulation of the Th17–Treg axis is an attractive therapeutic option that has been exploited both experimentally and more recently in early clinical trials.55 In order to prove their in vivo capacity, humanised mouse models have been developed, with the reconstitution of immunodeficient mice with human immune cells. Results from these studies have shown promise, with significant reduction in vasculopathy in an incorporated human aortic graft when mice were reconstituted with peripheral blood mononuclear cells (PBMCs) with co-transfer of expanded Tregs compared with PBMCs alone.56 Recently similar strategies have been introduced into haemopoietic stem cell transplantation, in which ex vivo expanded Tregs were administered following bone marrow transplantation, with evidence to suggest a reduction in the incidence of graft versus host disease.57 A European trial has recently begun to evaluate the safety of various immunomodulatory cell products in living donor kidney transplantation, and this includes administration of nTregs (the ONE study). The major risk of all cell therapies is the potential for plasticity, in which a protective regulatory cell differentiates into a pathogenic effector in vivo.
Immunology of graft rejection
Basic concepts and nomenclature of immunology
Recognition of danger
Histocompatibility
Major histocompatibility complexes
Assembly of the MHC–peptide complex
Other histocompatibility genes in rejection
Early inflammatory response
Ischaemia–reperfusion injury
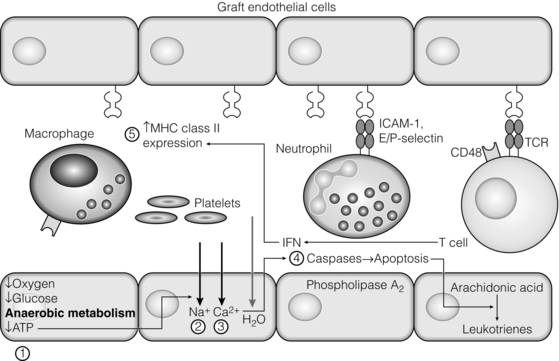
Ischaemic injury
Reperfusion injury
Sterile inflammation
Adaptive immune response to IRI
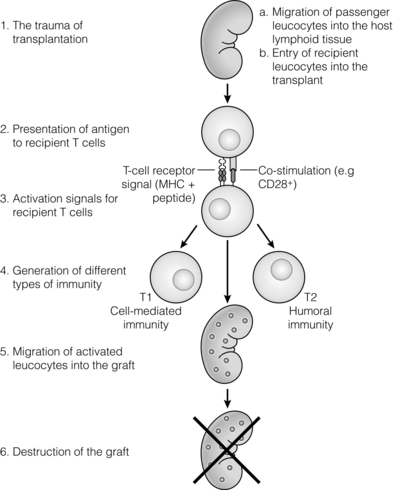
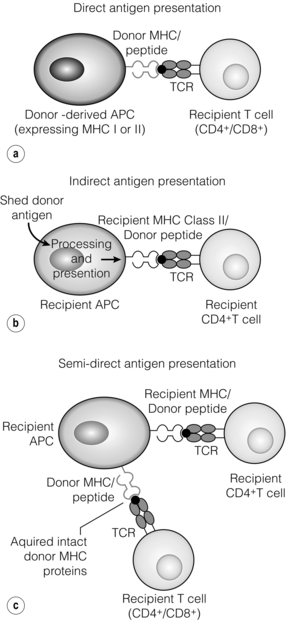
The alloimmune response
Direct allorecognition
Indirect allorecognition
Semi-direct allorecognition
Co-stimulation
Co-inhibitory molecules
TCR signalling
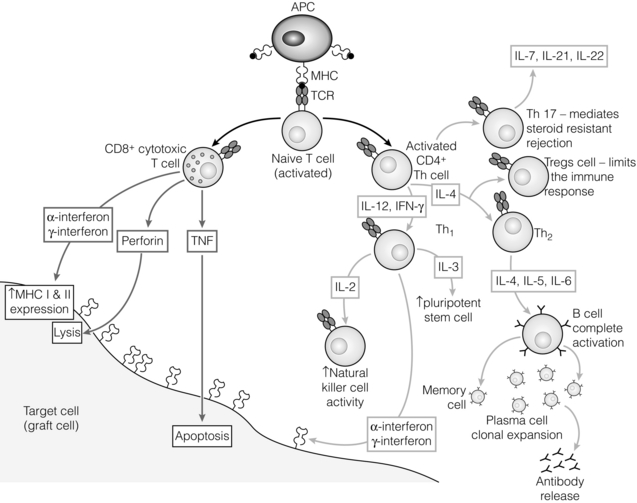
T-cell differentiation: the role of cytokines
T-cell responses
T helper 17 (Th17) response
T regulatory (Treg) response
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Immunology of graft rejection
Only gold members can continue reading. Log In or Register to continue