Immunobiologicals, Cytokines, and Growth Factors in Dermatology: Introduction
|
Immunobiologicals are compounds synthesized in living organisms that exhibit immune modulatory properties. They consist of recombinant cytokines, growth factors, antibody-based agents, and fusion proteins. Those with dermatologic indications are discussed in this chapter and are listed in Table 234-1. Fig. 234-1 illustrates the molecular targets of these compounds. Fig. 234-2 depicts the cellular events that mediate cutaneous inflammation and identifies the molecular interactions affected by specific immunobiological agents.
Action | Dosing for Dermatologic Indications | |
|---|---|---|
Recombinant interferons | ||
Interferon-α and Interferon-γ |
|
|
Recombinant Interleukins and growth factors | ||
Interleukin 2 |
|
|
Interleukin 1 receptor antagonist |
|
|
Granulocyte-macrophage colony-stimulating factor |
|
|
Platelet-derived growth factor |
|
|
Antibody-based therapeutics | ||
Anti-TNF-α |
|
|
Anti-IL12/IL23 |
|
|
Anti-LFA-1 |
|
|
Anti-CD20 |
|
|
Fusion proteins | ||
Alefacept |
|
|
Etanercept |
|
|
Denileukin Diftitox |
|
|
Figure 234-1
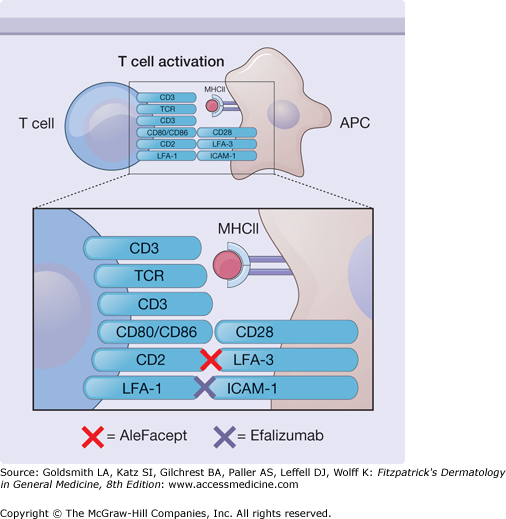
Immunobiologicals may modify immune responses via inhibition of T-cell activation. Efalizumab blocks interactions between leukocyte function-associated antigen 1 (LFA-1) and intercellular adhesion molecule 1 (ICAM-1), whereas alefacept interferes with interactions between CD2 and LFA-3. MHC II = major histocompatibility complex II; TCR = T−cell receptor.
Figure 234-2
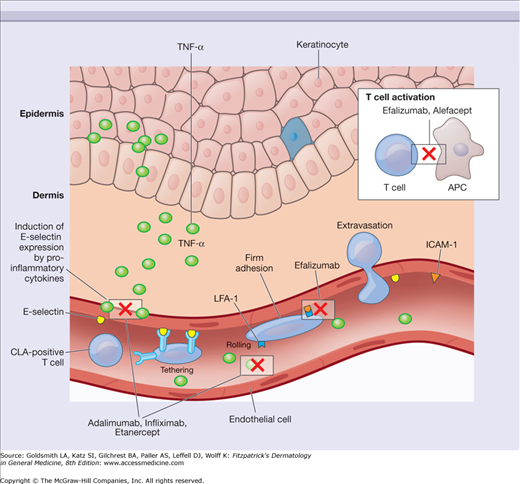
Cutaneous inflammation results in the production of inflammatory cytokines that induce keratinocyte proliferation and increase expression of molecules such as E-selectin. Immunobiologicals may interfere with immune cell trafficking by binding to specific cell surface receptors, deplete target cell populations through direct binding to cell surface proteins, suppress cytokine activity through the action of cytokine targeting compounds, and alter immune responses via the activity of recombinant cytokines with inhibitory or proinflammatory properties. The cellular interactions and cytokines targeted by several of the immunobiological agents discussed in this chapter are depicted. CLA = cutaneous lymphocyte-associated antigen; ICAM-1 = intercellular adhesion molecule 1; LFA-1 = leukocyte function-associated antigen 1; TNF-α = tumor necrosis factor-α.
Recombinant Cytokines and Growth Factors
Cytokines are low-molecular-weight polypeptides that exhibit paracrine and/or autocrine activity in the mediation of immune responses. They may act as growth factors by inducing the proliferation of specific immune cell populations. A particular cytokine may also influence the production of other cytokines and the behavior of cellular populations that express its receptor. Over the past two decades, recombinant cytokines have proven to be invaluable for the treatment of conditions in which immunologic aberrations exist or among which some clinical benefit might be derived from augmentation or suppression of the host immune response.
(See also Chapters 231 and 235)
IFN-α is a Type I interferon produced by plasmacytoid dendritic cells. It enhances cell-mediated cytotoxicity against viral disease and malignancy by increasing MHC I expression by antigen-presenting cells, stimulating NK cell activity, promoting the development of Th1 cells, and suppressing the production of Th2 cytokines.1,2
Recombinant IFN-α may be administered as a subcutaneous or intramuscular injection and has been utilized for a broad range of dermatologic conditions that include cutaneous T-cell lymphoma, melanoma, nonmelanoma skin cancer, Kaposi sarcoma, Behcet disease, hemangiomas, condyloma acuminatum, verruca vulgaris, and keloids.3 A pegylated form (containing polyethylene glycol) of this compound is currently available which has a longer half-life and increased patient tolerability.2 Prior to the availability of pegylated IFN-α, treatment was typically administered 3×/week with dosing dependent upon the condition being treated and patient response to therapy. Pegylated IFN-α may be administered once weekly with a steady state plasma level achieved within 5–9 weeks.4
There is extensive experience using IFN-α for the treatment of cutaneous T-cell lymphoma (CTCL). Objective clinical responses have been noted among 50%–75% of CTCL (Stage Ia–IVa) patients treated with different IFN-α dosing regimens.5 Typically, it is administered subcutaneously at a starting dose of 1.5–3.0 million units three times per week. When combined with other immunodulatory therapies, high clinical responses have been achieved among patients with Sézary syndrome.6
The most common side effect associated with IFN-α therapy is “flu-like” symptoms characterized by fever, sweats, chills, myalgias, and arthralgias. These symptoms typically resolve over the first ten days of therapy and can be managed with acetaminophen. Patients may also experience injection site reactions. Other associated side effects include fatigue, depression, weight loss, photosensitivity, peripheral neuropathy, psychosis, hypothyroidism, and sexual dysfunction.2,7 Laboratory abnormalities include elevated hepatic transaminases and leukopenia. Liver function tests and a complete blood count should be evaluated every 1 to 2 weeks upon initiation of therapy, or with any increase in dosing.2
IFN-α is contraindicated for use in patients with a known hypersensitivity to the drug or any of its components. Relative contraindications include a history of cardiovascular disease, renal disease, hepatic disease, central nervous system disorders, and/or preexisting mental illness. IFN-α is a pregnancy category C medication with unknown safety during lactation. Multiple case reports have suggested that IFN-α may induce, exacerbate, or unmask autoimmune disorders including immune-mediated thyroid disease, anemia, thrombocytopenia, sarcoidosis, and connective tissue disorders. IFN-α should be used with caution among patients with CD8+ CTCL, as multiple cases of disease progression have been reported in this setting.8–10 This finding may be explained by the inherent ability of Th1 cytokines to augment CD8+ T-cell activity.
IFN-γ is a cytokine produced by Th1 lymphocytes. Similar to IFN-α, it enhances cell-mediated cytotoxicity and suppresses Th2 cytokine production. IFN-γ has also been shown to enhance MHC 2 expression and interleukin 12 (IL12) production by antigen-presenting cells. In the dermatologic setting, it is reported to exhibit clinical efficacy in the management of chronic granulomatous disease and CTCL.
Children with chronic granulomatous disease treated with IFN-γ exhibited a significant reduction in the frequency of infections and associated hospitalizations.11 This has been attributed to its ability to partially correct defects in oxidative metabolism and promote superoxide release, thus enhancing phagocytes’ ability to combat infection.12
Studies regarding the use of IFN-γ in the treatment of CTCL are limited. Partial clinical responses have been reported among patients treated with IFN-γ during all stages of disease.13 This has been attributed, in part, to its ability to suppress the production of Th2 cytokines by malignant cells.14 Thus, IFN-γ offers an alternative therapeutic option for patients who may not tolerate or respond to IFN-α therapy secondary to the formation of neutralizing antibodies, cognitive dysfunction and/or fatigue—which are more frequently associated with IFN-α therapy.15
IFN-γ is administered via intravenous, intramuscular, and subcutaneous routes. The recommended dosing for children with chronic granulomatous disease is 50 μg/m2 three times a week for life.16 We have utilized IFN-γ doses ranging from 50–100 μg three times a week for the treatment of adult CTCL patients.2
Adverse effects associated with IFN-γ are similar to those reported for IFN-α and include flulike symptoms, elevated hepatic transaminases, leukopenia, and thrombocytopenia. IFN-γ is a pregnancy category C medication with unknown safety during lactation. Its contraindications are similar to those reported for FN-α.
Interleukin 2 (IL2) is a Th1 cytokine with antitumor activity produced by CD4+ lymphocytes in response to activation by antigen-presenting cells. IL2 has been shown to promote the proliferation and maintenance of helper T-cell populations. In addition, it has been shown to enhance both natural killer cell cytotoxicity and lymphokine-activated cell activity.17 In the dermatologic setting, recombinant human IL2 has been used for the treatment of melanoma.
Several studies have reported durable clinical responses among melanoma patients treated with IL2 therapy. This finding led to the FDA approval of this medication for the treatment of patients with metastatic disease. In early studies, high dose intravenous IL2 therapy (600,000–720,000 IU/kg) led to a 15% to 20% overall response among melanoma patients, with 4% to 6% achieving a complete clinical response.18–20 Unfortunately, prolonged responses are only achieved by a minority of patients. Over the past two decades, IL2 has been combined with other treatment modalities in an attempt to enhance clinical response rates and overall survival among melanoma patients.21
Adverse effects associated with infused IL2 include fever, chills, hypotension, thrombocytopenia, vascular leak syndrome, pulmonary edema, cardiac arrhythmias, renal compromise, and exfoliative erythroderma.2,3 IL2 is contraindicated for use in patients with a known hypersensitivity to the drug or any of its components. It is also contraindicated for patients with a history of an abnormal thallium stress test, abnormal pulmonary function tests, or organ allograft transplantation. IL2 is a pregnancy category C medication with unknown safety during lactation.
Interleukin 1 (IL1) is a proinflammatory cytokine produced by monocytes/macrophages that are involved in the recruitment of leukocytes to inflamed tissue, the generation of acute phase reactants, activation of osteoclast activity, induction of collagenase production by chondrocytes, and fever induction. In combination with other inflammatory cytokines, IL1 has been implicated in the pathogenesis of conditions that include rheumatoid arthritis and neonatal-onset multisystem inflammatory disease (NOMID).
The potentially damaging effects of prolonged IL1-α and/or IL1-β exposure are normally balanced by a naturally produced interleukin-1 receptor antagonist (IL1ra), which counters the effects of excessive IL1 exposure. If this protective mechanism is overcome, tissue damage may ensue. Anakinra is a recombinant IL1ra that was approved by the Food and Drugs Administration (FDA) in 2001 for the treatment of moderate-to-severe rheumatoid arthritis among adult patients who failed to respond to other antirheumatic drugs. It binds to the IL1 Type 1 receptor and competitively inhibits IL1 binding. The interaction between anakinra and the IL1 receptor does not activate the IL1 receptor accessory protein, thus no signal transduction cascade ensues.
Aside from adult rheumatoid arthritis, anakinra has been reported to exhibit clinical efficacy in the management of other inflammatory conditions, including Still disease, polyarticular juvenile rheumatoid arthritis, Muckle-Wells syndrome, and NOMID. This is a chronic inflammatory condition which presents during early childhood with a constellation of clinical findings that include an urticarial cutaneous eruption, central nervous system abnormalities, and bone deformities. A recent study to address the efficacy of anakinra in the management of NOMID reported a rapid clinical response to daily injections (1–2 mg/kg) with all patients achieving marked clearing of their cutaneous disease.22 A small study to address the long-term safety and efficacy of anakinra among NOMID patients reported sustained clinical improvement for 26–42 months (the duration of follow-up) with no adverse events aside from mild injection-site reactions.23
Clinical trials have suggested a small increased risk for infection among anakinra treated patients. Thus, treatment should not be initiated among patients with an active infection. The use of this drug in combination with TNF-inhibitor therapy should be avoided. Anakinra is contraindicated among patients with known allergies to Escherichia coli-derived proteins. The most common side effect associated with treatment is injection site reaction.
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a growth factor that induces the proliferation of myeloid precursors and is involved in the proliferation, differentiation, and migration of dendritic cells.24 In the dermatologic setting, GM-CSF has been reported to exhibit efficacy as a wound-healing agent by promoting the proliferation of keratinocytes in the setting of ulcerated, diseased skin.25,26 It has also been utilized as an antitumor agent in the treatment of melanoma and CTCL.
Regression of in-transit melanoma and cutaneous metastases have been reported with the administration of intralesional recombinant GM-CSF.27,28 A phase II trial in which GM-CSF was administered to postsurgical, clinically disease-free, stage III or IV melanoma patients reported enhanced survival among treated patients compared to matched historical controls.29 It has also been used as an adjuvant in combination with other treatment modalities for the treatment of melanoma.30,31
GM-CSF has been shown to induce the differentiation and proliferation of antigen-presenting cells,32 and may contribute toward disease improvement in CTCL through its ability to replete antigen-presenting cell populations, which are reduced in Sézary syndrome patients. Recombinant human GM-CSF has been used as adjuvant therapy in the treatment of patients with Sézary syndrome. Clinical improvement was reported in a patient treated with an aerosolized form of the medicine.33 Recombinant human GM-CSF dosing for the treatment of dermatologic disease has not been standardized. GM-CSF was used in combination with other immune modulating therapies for the treatment of Sézary syndrome leading to enhanced clinical responses in a non-placebo controlled study.6 Associated side effects include fatigue, myalgia, injection site reactions, and generalized cutaneous reactions. GM-CSF is contraindicated for use in patients with a known hypersensitivity to the drug or any of its components. Administration of this drug concurrently with chemotherapy is not recommended, as some combinations may increase myelosuppression. GM-CSF is a pregnancy category C medication with unknown safety during lactation.
(See also Chapter 235)
Platelet-derived growth factor (PDGF) is a peptide produced by platelets, macrophages, neutrophils, and smooth muscle cells that has proven to be of clinical benefit for the management of chronic wounds. It consists of two polypeptide chains (A and B) that form dimers linked by disulfide bonds. The PDGF-BB homodimer has been shown to promote granulation tissue formation, wound angiogenesis, re-epithelialization, and the proliferation of fibroblasts and smooth muscle cells in the cutaneous microenvironment. It is currently the only growth factor approved by the FDA for the treatment of diabetic foot ulcers.
Several randomized control studies have reported the efficacy of PDGF-BB for the treatment of pressure ulcers and lower extremity ulcers in diabetic patients. A phase III study reported significant efficacy of PDGF-BB in the time required to achieve complete wound closure for the treatment of chronic diabetic neuropathic ulcers of the lower extremities.34 This multicenter, double-blind, placebo-controlled study reported a 43% increase in the incidence of wound closure, and a 32% reduction in the time required to achieve complete wound closure compared to patients receiving a placebo gel. When combined with good wound care, daily application of PDGF has been found to substantially improve time to healing of chronic diabetic neuropathic foot ulcers.35
PDGF-BB (100 μg/g) is distributed as a topical gel that should be applied daily and refrigerated when not in use. Studies addressing the safety of recombinant human PDGF-BB found no increased risk for cardiovascular, respiratory, musculoskeletal, or central nervous system disorders among patients with lower extremity diabetic neuropathic ulcers treated with topical PDGF-BB compared to patients treated with placebo or general ulcer care.36 Patients receiving PDGF-BB therapy did not develop neutralizing antibodies to the drug, nor did they exhibit an increase in mortality relative to placebo. Cutaneous eruptions have been reported but are uncommon. Recombinant PDGF-BB is contraindicated for use in patients with a known hypersensitivity to the drug or any of its components. It also should not be applied to sites affected by cutaneous neoplastic disease. PDGF-BB is a pregnancy Category C medication with unknown safety during lactation. In 2008, a black box warning was added to the safety labeling of this drug as it was associated with an increase in cancer mortality among patients treated with three or more tubes (15 g/tube) of the gel.
Antibody-Based Therapeutics
Advances in immunobiology have led to our ability to synthesize antibodies that target specific cytokines and cell surface receptors. By utilizing them to interrupt specific signaling cascades, we have been able to manipulate immune responses through the activation or suppression of pathways mediating inflammation, tolerance, and antitumor immunity. The structural nature of a recombinant antibody can be determined by the suffix associated with its name [-ximab: chimeric monoclonal antibody (a hybrid antibody consisting of human and murine antibody components), -zumab: humanized monoclonal antibody, –umab: human monoclonal antibody, –cept: antibody fusion protein that mimics immunoglobulin].
(See Chapter 11)
Antitumor necrosis factor-α (anti-TNF-α) is a cytokine that has been linked to the pathogenesis of psoriasis and other inflammatory conditions including psoriatic arthritis, rheumatoid arthritis, and inflammatory bowel disease. It is produced by multiple cell types in the skin (keratinocytes, Langerhans cells, and dermal mast cells),37 which are believed to contribute to the pathogenesis of inflammatory dermatoses (see Fig. 234-2). TNF triggers its effects by binding to its p55 and p75 receptors expressed primarily on the surface of keratinocytes, neutrophils, endothelial cells, and fibroblasts. In combination with other proinflammatory cytokines, it is believed to be involved in the recruitment of immune cells to the cutaneous microenvironment.38 TNF may also contribute to both the inhibition of keratinocyte apoptosis and the induction of keratinocyte proliferation in psoriasis.39,40
Psoriasis is a chronic inflammatory condition of the skin that affects over 2% of the adult population.41 Elevated levels of TNF-α have been detected in the cutaneous lesions of psoriasis patients.42 Murine studies support a role for TNF-α in the activation and proliferation of lymphocytes, which induce psoriatic lesions.43 Elevated TNF-α levels have also been reported in patient serum and has been shown to correlate with disease activity.44,45
Three biologics, which inhibit TNF-α are approved for psoriasis: (1) infliximab, (2) adalimumab, and (3) etanercept. Infliximab and adalimumab are antibody-based therapeutics, whereas, etanercept is a fusion protein. Although all three drugs inhibit TNF-α, they are structurally different and have unique pharmacodynamic and pharmacokinetic profiles. Given their shared molecular target, certain of their properties will be discussed together as a “class” for comparative purposes in this section. Etanercept will be reviewed in more detail in the Section “Fusion Proteins.”
Infliximab and adalimumab bind to both soluble and membrane-bound TNF, whereas etanercept binds primarily to soluble TNF.46 Infliximab and adalimumab also have a greater propensity to induce lymphocyte apoptosis compared to etanercept, as they can lyse cells with membrane-bound TNF through complement activation and/or antibody-dependent, cell-mediated cytotoxicity.47 Given its inability to effectively associate with membrane-bound TNF, etanercept is incapable of activating complement-mediated apoptotic pathways. In terms of pharmacokinetics, the subcutaneous administration of adalimumab and etanercept yields smooth and uniform concentration time profiles at steady state, whereas the intravenous dosing of infliximab results in very high peak-to-trough ratios.48 Infliximab binds TNF quickly and irreversibly, whereas etanercept sheds about 50% of soluble TNF within 10 minutes of binding.47 The pharmacodynamic, pharmacokinetic, and structural differences between TNF inhibitors may result in differing safety and efficacy profiles. While infliximab appears to be more efficacious than etanercept in the management of autoimmune granulomatous diseases such as Crohn disease, sarcoidosis, and granulomatosis with polyangiitis (Wegener’s); its use47 has been associated with a higher risk of granulomatous infections.49
Etancercept, infliximab, and adalimumab are FDA approved for the treatment of moderate/severe plaque-stage psoriasis and psoriatic arthritis. Additionally, etanercept is FDA approved for rheumatoid arthritis, ankylosing spondylitis, and juvenile rheumatoid arthritis; infliximab is approved for rheumatoid arthritis, Crohn disease, ulcerative colitis, and ankylosing spondylitis; and adalimumab is approved for rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing spondylitis, and Crohn disease. Other TNF inhibitors, without dermatologic indications, have also been approved by the FDA and include golimumab (for the treatment of rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis) and certolizumab pegol (for the treatment of Crohn disease and rheumatoid arthritis).
Case reports and small case series suggest potential efficacy of TNF-α inhibitors for the management of numerous inflammatory dermatologic conditions that include apthous stomatitis, Behcet disease, cicatricial pemphigoid, dermatomyositis, eosinophilic fasciitis, hidradenitis suppurativa (HS), multicentric reticulohistiocytosis, necrobiosis lipoidica diabeticorum, pityriasis rubra pilaris, pyoderma gangrenosum, sarcoidosis, scleroderma, Sneddon-Wilkinson syndrome, and SAPHO (synovitis, acne, pustulosis, hyperostosis, and osteitis) syndrome.50 A phase 2, randomized, placebo-controlled trial conducted at a single institution reported a higher response rate among HS patients treated with infliximab (5 mg/kg on weeks 0, 2, and 6) compared to placebo. More specifically, a greater percentage of infliximab-treated patients achieved a >50% reduction in baseline HS Severity Index (HSSI) score at week 8 compared to placebo.51 In addition, 60% of infliximab-treated patients achieved a 25% to 50% reduction in HSSI compared to 6% of placebo.
TNF inhibitors have been the most extensively studied class of biologics and therefore they have detailed and relatively robust safety data compared to other biologics. TNF inhibitors are immunosuppressive, and therefore, patients should be carefully screened for signs and symptoms of malignancy and infection prior to initiating therapy as well as during the course of treatment. Product labeling and published guidelines recommend screening for latent tuberculosis infection with a tuberculin skin test prior to initiating therapy with etanercept, infliximab, and adalimumab.52 A whole blood test, such as QuantiFERON-TB gold or T-SPOT.TB assay, is an alternative screening method that offers higher specificity and comparable sensitivity to the traditional tuberculin skin test. It is particularly useful among BCG-vaccinated individuals who are more likely to exhibit a false-positive PPD reaction. TNF inhibitors should be used with caution in patients with a history of congestive heart failure—infliximab at doses >5 mg/kg is contraindicated in patients with moderate-to-severe congestive heart failure.
Infliximab may be associated with liver function test abnormalities, which, in rare cases, have led to hepatic failure. Additionally, TNF inhibitors have been associated with reactivation of hepatitis B in patients who are chronic carriers. Some of these cases have been fatal; therefore, one should consider monitoring liver function tests during TNF therapy and screening at-risk patients for hepatitis B. Small cases series suggest that TNF inhibitors can be used safely in patients with Hepatitis B when lamivudine is given concurrently. Larger studies, however, will be necessary to further substantiate these findings.53 No exacerbation of liver disease has been reported among hepatitis C-positive patients receiving TNF inhibitor therapy. In fact, it has been suggested that TNF inhibitor therapy may contribute toward improving clinical outcomes among this patient population given their potential to suppress TNF-induced hepatic inflammation and fibrosis.54,55 A recent multicenter study addressing the efficacy of etanercept in the treatment of alcoholic hepatitis reported increased mortality among patients with moderate-to-severe disease after 6 months of therapy; thus, the use of TNF inhibitors in this setting should be avoided if possible.56
TNF inhibitors are associated with serious infections including pneumonia, sepsis, tuberculosis, histoplasmosis, coccidioidomycosis, and other invasive fungal infections, thus accounting for the black box warning for infection. Product labeling also indicates that for patients who have resided in regions where histoplasmosis and coccidioidomycosis are endemic, the risks and benefits of TNF inhibitor therapy should be carefully considered. A recent meta-analysis of rheumatoid arthritis patients treated with infliximab or adalimumab in randomized controlled trials suggested that use of these agents result in one excess serious infection for every 59 patients treated for a period of 3–12 months.57 Use of etanercept with anakinra has been associated with an increased risk of serious infections, and therefore, it is recommended that anakinra not be used concurrently with any TNF inhibitor. One prospective study reported an increased incidence of herpes zoster among rheumatoid arthritis patients treated with anti-TNF monoclonal antibodies (adalimumab, infliximab) compared to conventional disease-modifying antirheumatic drugs (DMARDs).58 A meta-analysis addressing the risk of serious infections (infection requiring antibiotic treatment and/or hospitalization) among rheumatoid arthritis patients treated with adalimumab and infliximab reported a significantly increased risk for such infections compared to placebo.57 Another meta-analysis addressing the risk of serious infections among RA patients treated with adalimumab, etanercept, or infliximab, with and without adjustment for exposure, reported no increased risk of serious infection at manufacturer recommended doses, however, there was a two-fold increased risk of serious infection among patients treated with higher doses (two to three times the recommended dose).59
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


