Hypomelanoses and Hypermelanoses: Introduction
|
An Algorithmic Approach to Pigmentation Disorders
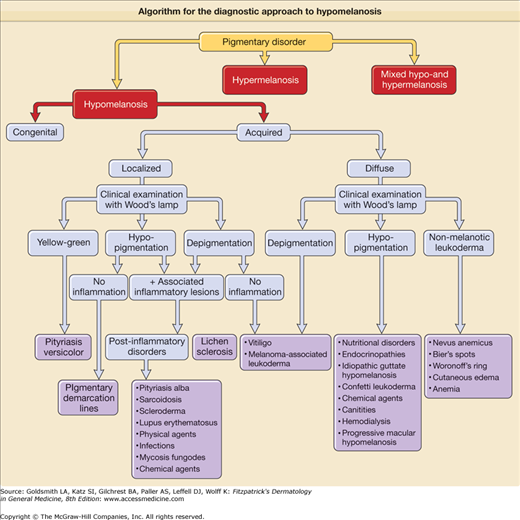
Pigmentation disorders of the skin can either be hypomelanotic, hypermelanotic, or may present with a pattern of mixed hypo- and hypermelanosis. The diagnosis of these disorders can be quite challenging. An algorithmic approach based on clinical features and history of pigmentary disorders is used throughout this chapter and serves as a guide for the clinician’ diagnosis and treatment (eFigs. 75-0.1 and 75-0.2) (Table 75-1, eTable 75-1.1).
Nevus of Ota | Nevus of Ito | Mongolian Spot | Nevus of Hori | Dermal Melanocyte Hamartoma | |
|---|---|---|---|---|---|
Epidemiology | • Mostly congenital • Sporadic (rare familial cases) • Asian and female predominance | • Mostly congenital • Sporadic (rare familial cases) • Asian and female predominance | • Congenital • Often familial • Asian, African, and Hispanic population with slight male predominance | • Acquired • Familial or sporadic • Asian and female predominance | • Congenital |
Clinical presentation | • Blue to slate-gray mottled macular hyperpigmentation | • Blue to slate-gray mottled macular hyperpigmentation | • Uniform blue to slate-gray macular hyperpigmentation | • Brown–blue progressing to slate-gray mottled macular hyperpigmentation | • Mottled hyperpigmentation with small blue–gray macules in a diffuse pigmented patch |
Distribution | • Trigeminal nerve | • Acromioclavicular nerve | • Lower back and sacrum | • Especially malar region of the cheek (also forehead, upper eyelids, temple) | • Dermatomal distribution |
Histology | • Spindle-shaped melanocytes diffusely throughout the dermal layers. Sometimes more band-like melanocytic proliferation and stromal fibrotic reaction. | • Spindle-shaped melanocytes diffusely throughout the dermal layers. Sometimes more band-like melanocytic proliferation and stromal fibrotic reaction | • Spindle-shaped melanocytes diffusely throughout the dermal layers | • Dermal melanocytes in the upper and middermis | • Dermal melanocytes in the upper two-thirds of the dermis (including subpapillary layer) |
Therapy | • Q-switched laser • Cryotherapy • Surgery | • Q-switched laser • Cryotherapy • Surgery | • Usually spontaneous regression during childhood | • Q-switched laser in combination with bleaching cream and chemical peels | • None |
Associated features | • Rare malignant transformation | • No associated features of medical concern | • Possible association with inborn errors of metabolism | • No associated features of medical concern | • None |
eFigure 75-0.2
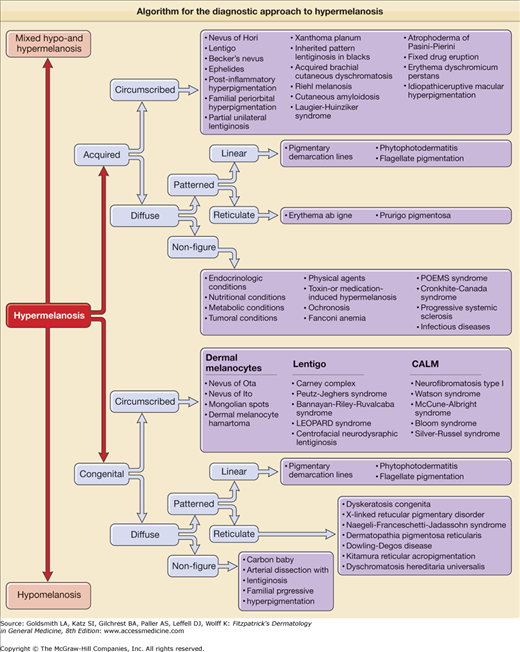
Algorithm for the diagnostic approach to hypermelanosis. CALM = café-au-lait macule; LEOPARD syndrome = lentigines; electrocardiogram conduction defects; ocular hypertelorism; pulmonary stenosis; abnormalities of genitalia; retardation of growth, and sensorineural deafness; POEMS = polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes.
Topical Treatments | Anti-Infectious Drugs | Central Nervous System Drugs | Antineoplastic Agents | Miscellaneous |
|---|---|---|---|---|
Aminolevulinic acid Carmustine (BCNU) Bergamot Bimatoprost Carteolol Chlorhexidine Hydroquinone Imiquimod Latanoprost Nitrogen mustard Tretinoin Verteporfin Antihypertensive drugs Acebutolol Betaxolol Bisoprolol Captopril Clonidine Diltiazem Esmolol Indapamide Labetalol Methyldopa Metoprolol Minoxidil Nisoldipine Propranolol Spironolactone Timolol Heavy metals Arsenic Bismuth Gold (compounds) Iron Lead Mercury Silver | Amphotericin B Ceftriaxone Chloroquine Cidofovir Clofazimine Dapsone Demeclocycline Doxycycline Emtricitabine Enoxacin Foscarnet Ganciclovir Grepafloxacin Griseofulvin Hydroxychloroquine Indinavir Ketoconazole Linezolid Lomefloxacin Minocycline Ofloxacin Oxytetracycline Pyrimethamine Quinacrine Quinine Ribavirin Rifabutin Rifapentine Saquinavir Sertaconazole Smallpox vaccine Sparfloxacin Sulfadiazine Terbinafine Tetracycline Voriconazole Zidovudine | Amitriptyline Carbamazepine Chlorpromazine Citalopram Clomipramine Clonazepam Desipramine Diazepam Donepezil Eletriptan Fluoxetine Fluphenazine Fluvoxamine Haloperidol Imipramine Kava Loxapine Mephenytoin Mesoridazine Methamphetamine Molindole Olanzapine Paroxetine Perphenazine Phenytoin Pimozide Prochlorperazine Promazine Promethazine Risperidone Ropinirole Thioridazine Thiothixene Tiagabine Tolcapone Topiramate Trifluoperazine Venlafaxine Zaleplon | Bevacizumab Bleomycin Busulfan Capecitabine Carboplatin Carmustine Cisplatin Cyclophosphamide Dactinomycin Daunorubicin Doxorubicin Epirubicin Estramustine Etoposide Floxuridine Fluorouracil Hydroxyurea Ifosfamide Irinotecan Mechlorethamine Mercaptopurine Methotrexate Mitomycin Mitotane Mitoxantrone Paclitaxel Pentostatin Procarbazine Thiotepa Vinblastine Vincristine Vinorelbine Hormones Chlorotrianisene Corticosteroids Diethylstilbestrol Estrogens Insulin Leuprolide Medroxyprogesterone Oral contraceptives Progestins Stanozolol | Alitretinoin Amiodarone Azathioprine Cetirizine Cevimeline Cyclobenzaprine Cyclosporine Deferoxamine Dicumarol Dinoprostone Etodolac Glatiramer Heroin Interferon Isotretinoin Ketoprofen Leflunomide Levobupivacaine Lidocaine Methimazole Methoxsalen Methysergide Metoclopramide Niacin Nicotine Orphenadrine Pantoprazole Pentazocine Phenazopyridine Phenolphthalein Propylthiouracil Psoralens Quinidine Rabeprazole Riluzole Sulfasalazine Tacrolimus Toremifene Trioxsalen Vitamin A |
Hypomelanosis
Generalized hypopigmentation, including the various defects, classified as “albinism” has been discussed in Chapters 73 and 74. Localized forms of hypomelanosis can be related to defects in melanocyte precursor migration and disordered pigment transfer between melanocytes and keratinocytes, and as a result of postinflammatory changes.
Some genetic disorders with reduced skin and hair pigmentation are caused by impaired melanocyte migration/differentiation or melanosome abnormalities.1 Piebaldism, Waardenburg syndrome, and Tietze syndrome, characterized by a localized absence of melanocytes resulting in “white-patch” patterns, belong to the first group whereas oculocutaneous albinism (OCA), Griscelli syndrome (GS), Elejalde syndrome (ES), Chédiak–Higashi syndrome (CHS), and Hermansky–Pudlak syndrome (HPS) belong to the second group. OCA, CHS, and HPS are discussed in Chapter 73; GS and ES are discussed here. They are rare autosomal recessive disorders with abnormal biogenesis or transport of “lysosome-related organelles” (a group of specialized cytoplasmic organelles including melanosomes, platelet dense bodies, and lymphocyte lytic granules).2 GS, ES, and CHS are termed “silvery hair syndromes” because hair of these patients has a particular silver–gray hue.3
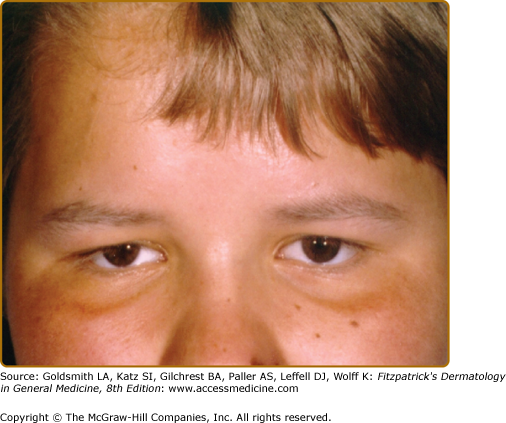
Three types of GS [GS types I–III (GS1–3)] are known. The phenotype of the patients is characterized by various degrees of skin hypopigmentation and hair with a silvery shine (Fig. 75-1), usually lighter than in unaffected family members. Furthermore, neurological signs and symptoms and/or immunologic impairment with “accelerated phases” of uncontrolled lymphocyte and macrophage activation with lymphohistiocytic infiltration of the central nervous system (CNS) are associated.4 This lymphoproliferative syndrome is similar to that observed in, for example, virus-associated hemophagocytic syndrome.5
Briefly, patients with GS type I (GS1) are characterized by primary and severe neurological symptoms occurring early in life or even at birth without signs of an accelerated phase. These symptoms can include seizures, spasticity, psychomotor retardation, peripheral facial palsy, hemiparesis, encephalopathy, and hypotonia. CNS disorder is pertinent and never regresses with time.
Immunological and hematological manifestations are only observed in GS type II (GS2) and include: anemia, neutropenia, and lack of natural killer cell function, with development of an accelerated phase of the disease with fever, jaundice, hepatosplenomegaly, lymphadenopathy, pancytopenia, and generalized lymphohistiocytic infiltrates of various organs, including the CNS. Onset of the accelerated phase seems to be associated with viral or bacterial infections. When a remission occurs, recurrent accelerated phases with increasing severity will be observed.
Neurological problems may also occur in GS2 patients and are related to lymphocyte infiltration of the CNS. Associated symptoms are, for example, hyperreflexia, seizures, signs of intracranial hypertension (e.g., vomiting), hypertonia, nystagmus, and ataxia. Psychomotor development is normal at onset, and regression of CNS signs, at least in part, can be observed during remission, although some sequelae may be irreversible. Skin hypopigmentation and silvery-grey hair in Griscelli patients are not caused by deficient melanosome biogenesis but by impaired intramelanocytic melanosome transport. Murine models with autosomal recessive mutations on the dilute (d), ashen (ash), and leaden (ln) locus present a phenotype close to that of their human GS counterparts.6 These loci respectively encode three molecules, myosin Va, Rab27a, and melanophilin (Mlph) that act as a tripartite complex linking the melanosome to subcortical actin. When MYO5A (GS1), RAB27A (GS2), or MLPH (GS3) is mutated in human melanocytes, the tripartite complex fails to form and melanosomes can no longer be tethered near the plasma membrane for transfer to keratinocytes, leading to a pigmentary defect.7
The prognosis for patients with GS is generally bad. They usually die in the first or second decade of their life if accelerated phases are not treated adequately. As for GS2 therapy, allogeneic bone marrow transplantation has appeared to be successful in some cases, especially when carried out at an early age.8–11 Palliative therapy consists of suppressing the accelerated phases with immunosuppressive therapy (high-dose corticosteroids, cyclosporine) and chemotherapeutic agents (methotrexate, etoposide, cytosine arabinoside). For GS1 there is no therapy. Palliation consists of treating infections with antibiotics.12
The hematologic, immunologic, and neurologic findings in GS1–3 presumably relate to organelle transport difficulties in the respective organ systems.
 Initially, the dilute mouse (d) phenotype model contributed to the elucidation of the first candidate gene for the Griscelli locus. In 1978, Griscelli et al13 described for the first time two unrelated patients with hypomelanosis, neurological impairment, and primary immunodeficiency, later known as the Griscelli–Prunieras syndrome. They suggested that the observed pigment dilution and neurological impairment resembled the phenotype of the d/d mouse (dilute).13 Mercer et al found that the myosin Va motor protein, which encodes an actin-driven motor protein of the myosin heavy chain family mapping to chromosome 9, was encoded by the mouse dilute locus.14
Initially, the dilute mouse (d) phenotype model contributed to the elucidation of the first candidate gene for the Griscelli locus. In 1978, Griscelli et al13 described for the first time two unrelated patients with hypomelanosis, neurological impairment, and primary immunodeficiency, later known as the Griscelli–Prunieras syndrome. They suggested that the observed pigment dilution and neurological impairment resembled the phenotype of the d/d mouse (dilute).13 Mercer et al found that the myosin Va motor protein, which encodes an actin-driven motor protein of the myosin heavy chain family mapping to chromosome 9, was encoded by the mouse dilute locus.14
 Mutations in the human MYO5A gene, on chromosome 15q21 were found in patients with Griscelli syndrome (GS). Basically, patients having GS1 display hypomelanosis and severe primary neurological impairment. Recently a form of GS1 was found of which the phenotype was restricted to hypopigmentation and associated with loss of exon F of MYO5A.15
Mutations in the human MYO5A gene, on chromosome 15q21 were found in patients with Griscelli syndrome (GS). Basically, patients having GS1 display hypomelanosis and severe primary neurological impairment. Recently a form of GS1 was found of which the phenotype was restricted to hypopigmentation and associated with loss of exon F of MYO5A.15
 The mouse ashen (ash) mutation is situated in the small guanine triphosphatase Rab27a gene located on chromosome 9.16 Defects were observed in mainly two types of lysosome-related organelles, (1) melanosomes in melanocytes and (2) lytic granules in cytotoxic T lymphocytes. In Griscelli patients, the majority of detected mutations are associated with the RAB27A gene.15,17,18 Rab27a is known to interact in its guanine triphosphate-bound form with the melanosome membrane through its posttranslational C20 geranylgeranyl lipid tail. There it acts as a receptor for myosin Va through an indirect interaction via the Rab27a effector melanophilin.19 Without Rab27a the tripartite complex cannot be formed and melanosome transport will be impaired. This second type of GS (GS2) is characterized by the same hypopigmentation as GS1 but is associated with an immune defect, leading to episodes of life-threatening uncontrolled T lymphocyte and macrophage activation known as hemophagocytic syndrome during which activated T cells and macrophages infiltrate various organs. Leukocyte infiltration in the brain may lead to secondary neurological impairment. Primary central nervous system (CNS) defects are not observed in GS2 patients.
The mouse ashen (ash) mutation is situated in the small guanine triphosphatase Rab27a gene located on chromosome 9.16 Defects were observed in mainly two types of lysosome-related organelles, (1) melanosomes in melanocytes and (2) lytic granules in cytotoxic T lymphocytes. In Griscelli patients, the majority of detected mutations are associated with the RAB27A gene.15,17,18 Rab27a is known to interact in its guanine triphosphate-bound form with the melanosome membrane through its posttranslational C20 geranylgeranyl lipid tail. There it acts as a receptor for myosin Va through an indirect interaction via the Rab27a effector melanophilin.19 Without Rab27a the tripartite complex cannot be formed and melanosome transport will be impaired. This second type of GS (GS2) is characterized by the same hypopigmentation as GS1 but is associated with an immune defect, leading to episodes of life-threatening uncontrolled T lymphocyte and macrophage activation known as hemophagocytic syndrome during which activated T cells and macrophages infiltrate various organs. Leukocyte infiltration in the brain may lead to secondary neurological impairment. Primary central nervous system (CNS) defects are not observed in GS2 patients.
 Mlph, located on chromosome 1 and encoding a member of the Rab effector family, was identified as the candidate gene in ln mice by positional cloning.20 Recently, Menasche and coworkers presented genetic and functional evidence that a third form of GS (GS3) resulted from mutation in the human MPLH gene, mapped to chromosome 2q37. A homozygous C103T transition was identified in exon 1 of the patient’s gene causes a putative R35W substitution. Interestingly, phenotypic expression of GS3 is restricted to the characteristic hypopigmentation of this syndrome.
Mlph, located on chromosome 1 and encoding a member of the Rab effector family, was identified as the candidate gene in ln mice by positional cloning.20 Recently, Menasche and coworkers presented genetic and functional evidence that a third form of GS (GS3) resulted from mutation in the human MPLH gene, mapped to chromosome 2q37. A homozygous C103T transition was identified in exon 1 of the patient’s gene causes a putative R35W substitution. Interestingly, phenotypic expression of GS3 is restricted to the characteristic hypopigmentation of this syndrome.
ES, also referred to as neuroectodermal melanolysosomal disease, is another autosomal recessive pigment mutation with silvery hair, pigment abnormalities, and severe CNS dysfunction21,22 similar to those of GS1. ES patients do not manifest the hemophagocytic syndrome or immunological impairment associated with GS2.23,24
Certain patients with ES have clinical and histologic features suggestive of GS1, indicating that these are other cases of MYO5A mutations.25,26 Further work will be required to define the molecular basis of ES and to group all patients with these rare disorders correctly.
(See Chapter 189).
Pityriasis versicolor is characterized by slightly scaling macules that can either be hypopigmented, pink or salmon-colored, or hyperpigmented (Fig. 75-2); variants with red and black macules have also been described. The prevalence of pityriasis versicolor is very high in hot and humid climates.
Figure 75-2
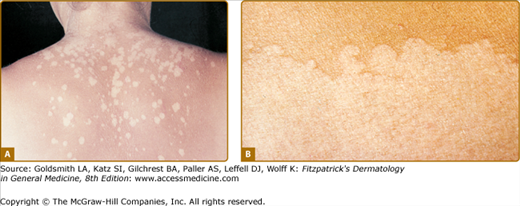
Pityriasis versicolor. A. Typical macules are round, very well circumscribed, have fine scale, and are off-white to tan colored. Typical distribution involves the upper back and upper chest. Involvement of the lower arms and legs and of the face is unusual. B. Confluent macules create scalloped borders. This is a characteristic pattern of macules of pityriasis versicolor.
This superficial mycosis is caused by Malassezia species of which M. globosa, M. sympodialis, and M. Furfur are most frequently identified in lesional scales. M. Furfur cultures produce a wide range of fluorochromes and pigments that appear to lead to depigmentation, high resistance to ultraviolet (UV)-induced tanning, and lack of inflammation observed in pityriasis versicolor.27 In approximately one out of three patients, a yellow–green fluorescence is visible using Wood’s light. It should be differentiated from the nonscaling depigmented lesions of vitiligo that frequently affect hands and feet, whereas pityriasis versicolor is mainly located on the trunk. The slightly scaling patches of pityriasis alba (PA) usually occur on the face and limbs and are nonfluorescent. Many local and systemic antifungal preparations are effective but relapses often occur.28,29
Originally described in Japanese patients as a line present on upper and lower extremities corresponding to a border of transition between the more deeply pigmented skin of the outer (dorsal) surfaces and the lighter inner (ventral) surfaces, the concept of pigmentary demarcation lines was expanded to include five specific patterns, labeled A–E, with A: upper anterolateral arms, across pectoral area (Fig. 75-3), B: posteromedial portion of lower limb, C: vertical hypopigmented line in pre- and parasternal area, D: posteromedial area of spine, and E: bilateral aspect of chest, marking from midthird of clavicle to periareolar skin. In a population survey of black and white patients it was determined that these lines appeared in early childhood. They are present in the majority of female black adults, with types A and B being most prevalent. Seventy-five percent of black adult males had at least one type of pigmentary demarcation line, with type C the most prevalent. Fifteen percent of white female adults had at least one line, and 14% of black women saw type B lines appearing during pregnancy.30 The lines of pigmentation are often called Futcher’s lines in the United States.
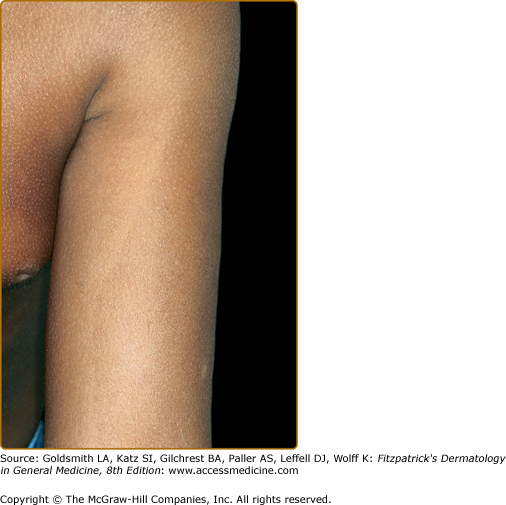
The pathogenesis of pigmentary demarcation lines is uncertain. Some authors believe that these lines coincide with lines of Blaschko and are secondary to a form of pigmentary mosaicism. Others suggest that there is no true genetic difference but hypothesize that the difference between cells on the dark, posterior side versus the light, anterior side are due to the normal function of genes, such as the agouti locus.31
According to some reports, pigmentary demarcation lines do not follow lines of Blaschko but have a pattern similar to lines of Voigt, which separate dermatomes arising from nonconsecutive dorsal roots. The difference in melanogenesis would be explained by a strict territorial control by different homeobox genes during development.32
PA is a common benign condition mainly affecting the head and neck region of preadolescent children. Although the disease is more noticed in darker skin types, there is no predilection for either sex or skin type. The etiology and pathogenesis remain poorly understood. PA is widely understood to represent mild atopic dermatitis. Unprotected sun exposure, frequent bathing, and hot baths are strongly related to the development of PA. Lower serum levels of copper, a cofactor for tyrosinase, could also play a role in the pathogenesis of this condition.
PA may present as a pink patch with an elevated border, fading after several weeks into a paler spot covered with powdery white scale (Fig. 75-4). The lesions progress to nonscaly hypopigmented macules persisting for months or years. The three stages may occur simultaneously.
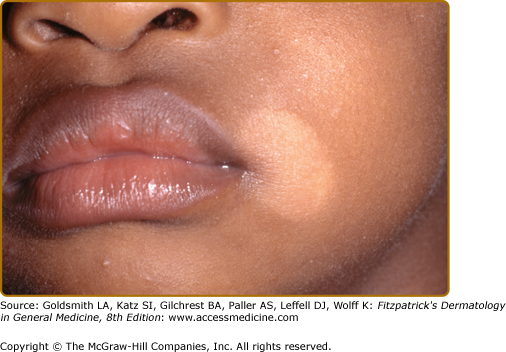
Histologically, there is markedly reduced pigment in the epidermis of lesional skin, but no significant difference in melanocyte count was found between lesional and normal skin. Ultrastructurally, degenerative changes in melanocytes and a reduced number of melanosmes within keratinocytes were seen.33 Extensive PA often also involves the inferior torso in a symmetric pattern. The lack of a preceding inflammatory phase and spongiosis differentiate extensive PA from the classic form. This particular form of PA may overlap with progressive macular hypomelanosis (PMH), a condition mainly described in young female adults, characterized by relapsing hypopigmented, nonscaling patches involving the back, particularly after summer.34
Pigmenting PA is a variant associating classic PA with a superficial dermatophyte infection, almost always affecting the face. It is clinically characterized by a bluish hyperpigmentation, attributed to melanin deposits in the dermis surrounded by a hypopigmented scaly area. One-third of patients have concurrent classic PA.
Differential diagnosis includes any localized form of hypopigmentation, especially inflammatory skin conditions associated with postinflammatory hypopigmentation, such as psoriasis, but also with fungal infection, nevus depigmentosus, nevus anemicus, tuberous sclerosis, mycosis fungoides (MF), or vitiligo.
The disease is self-limited and treatment is often not completely successful. Topical steroids and emollients are helpful. Topical tretinoin has also been used with success, but may be irritating. Extensive PA and pigmenting PA have also responded to UV therapy and oral antifungals, respectively. Supportive measures such as decreasing sun exposure, use of sunscreens, and reducing frequency and temperature of baths should be recommended.
(See Chapter 152).
Hypopigmentation is a rare manifestation of sarcoidosis. Hypopigmented macular lesions scattered over the trunk and extremities but also papular or nodular lesions may be present. The presence of noncaseating dermal granulomas, usually most evident in biopsies of indurated lesions, reinforce the diagnosis. A reduction in melanin content of the epidermis with preservation of melanocytes has been demonstrated.
(See Chapter 157).
Hypopigmentation has been described as a pigmentary change in morphea (localized scleroderma) (Fig. 75-5) and scleroderma (progressive systemic sclerosis). Localized hypopigmentation and/or hyperpigmentation are seen in areas of localized sclerosis. Focal depigmentation with perifollicular hyperpigmentation (“salt and pepper pigmentation”) especially on upper trunk and extremities, mimicking vitiligo, is reported in up to 30% of patients with scleroderma. The coexistence of scleroderma/morphea and vitiligo has also been reported.42–44
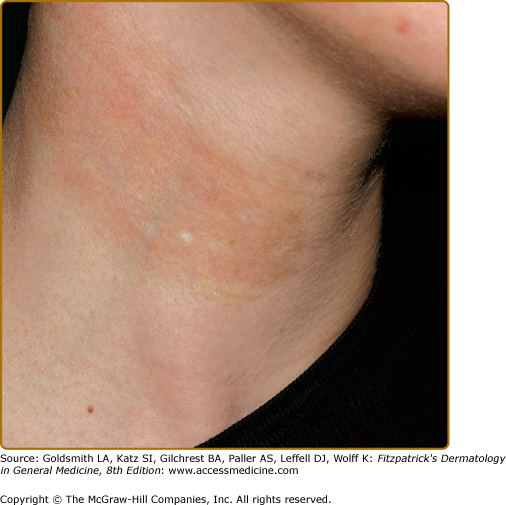
(See Chapter 155).
Pigment alterations are frequently seen in discoid lupus erythematosus. Hypopigmented patches result from interface dermatitis with destruction of the epidermal basal layer containing melanocytes. “Burned out” lesions are atrophic and depigmented and may be surrounded by hyperpigmentation. Cutaneous depigmentation is also reported in systemic lupus erythematosus, usually localized to inflammatory skin lesions. Biopsy specimens in depigmented skin feature degeneration of the basal layer with epidermal atrophy, a variable number of melanocytes, and pigmentary incontinence in the superficial dermis. The mechanism of hypopigmentation in lupus is not known but could be postinflammatory or cicatricial.
Vitiligo has also been reported in association with lupus erythematosus.45 A shared genetic predisposition may explain the association of these two autoimmune disorders.46
(See Chapter 146).
MF, the most common type of cutaneous T-cell lymphoma, is clinically characterized by three cutaneous phases: (1) the patch phase, (2) the plaque phase, and (3) the tumor phase. Several pigmentary changes have been described in MF.
In poikiloderma vasculare atrophicans, a variant of the patch stage, mottled pigmentation, atrophy and telangiectasia of the involved skin are observed. A mix of hyper- and hypopigmentation may remain after regression of the skin lesions following treatment.
Hypopigmented MF is an uncommon variant of this lymphoma. It mainly develops before the fourth decade, predominantly in juvenile-onset cases and in dark-skinned individuals, without sex predilection. Irregular hypopigmented patches with variably distinct borders are preferentially located on trunk and extremities. Erythema, scaling, and infiltration may be present. A central area of normal pigmentation may be observed. These lesions may also be associated with the more typical lesions of the three cutaneous phases. The hypopigmentation develops without preceding skin changes and occasionally complete depigmentation is observed.
Histopathologically, hypopigmented MF is characterized by minimal dermal involvement, lack of epidermal atrophy, and moderate to marked exocytosis. Pigment incontinence and decrease or absence of melanin may be observed. Infiltrating lymphocytes often have a T-suppressor cell CD8+ phenotype, but a CD4+ phenotype has also been reported.
Electron microscopy may disclose degenerative changes in melanocytes. Melanocytes may be incompletely melanized or occasionally reduced in number. The number of melanosomes within keratinocytes is normal or decreased and melanin-containing macrophages can be observed in the papillary dermis. T-cell receptor gene rearrangement analysis may help to confirm the diagnosis but is often negative, as in early stage MF.
The pathogenesis of hypopigmented MF is not clear. At least in cases showing a CD8+ phenotype, hypopigmentation could be due to the melanocytotoxic effect of nonneoplastic CD8+ lymphocytes, as hypothesized in vitiligo.
Hypopigmented MF responds well to treatment, particularly psoralen and UVA light (PUVA) or narrowband UVB therapy. It has a relatively benign course, although recurrences are common. Hypopigmented MF should be distinguished from other causes of diffuse hypopigmentation, especially vitiligo, tinea versicolor, PA, or postinflammatory hypopigmentation.47–51
Nonvenereal treponematoses are currently endemic in parts of Central and South America, Africa, Asia, and the Pacific Islands and can be severely disfiguring. Depigmentation is seen in several stages of yaws, bejel, and pinta. When the primary lesion of yaws disappears, a typical depigmented and pitted scar remains. In the tertiary stage of bejel, gummatous nodules develop in the skin and in other organs. Most skin lesions regress, leaving depigmented, noncontracted scars. Pinta is the only treponematosis that only affects the skin and causes pigmentary abnormalities in the first, second, and third stages of the disease. The sentinel lesion of pinta may heal at the end of the primary stage, leaving a macular dyschromia. The secondary stage is characterized by the appearance of the so-called pintids, which are initially red but often turn brown, slate-blue, gray, or black. This stage can last several years, leading to a mix of depigmentation and hyperpigmented lesions. Generalized pigmentary abnormalities develop during the tertiary stage. A symmetrical pattern of vitiligo-like lesions and brown, gray or blue and black lesions is seen over bony prominences (Fig. 75-6).52
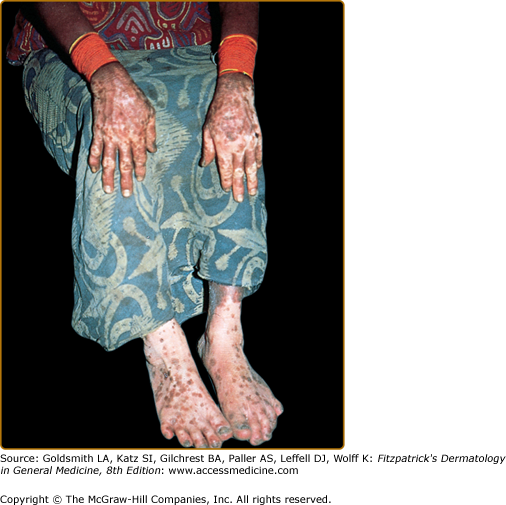
(See Chapter 207).
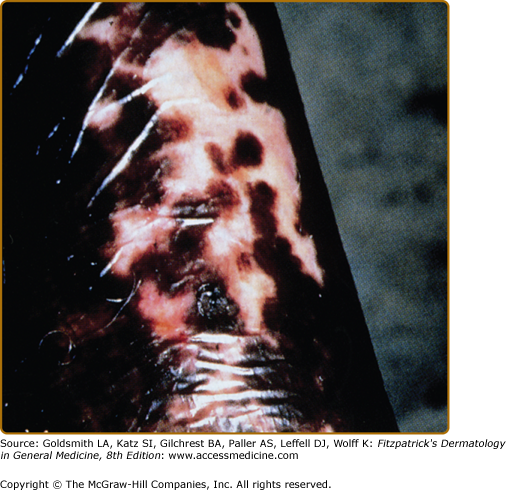
Several different skin manifestations can become apparent during the course of onchocerciasis. Onchocercal depigmentation or “leopard skin” is rarely associated with itch and is one of the most common skin manifestations of onchocerciasis. Hypopigmented patches with perifollicular spots of normally pigmented skin, typically occur symmetrically on the pretibial area of older people in endemic areas.53 (Fig. 75-7).
(See Chapter 206).
Post-kala-azar dermal leishmaniasis develops in insufficiently treated kala-azar or visceral leishmaniasis, which is caused by Leishmani. donovani. Skin manifestations of PKDL are nodules and plaques, facial erythema, and hypopigmented macules. Nodules and plaques typically develop around the mouth and spread to the face, arms and chest, but the macules may occur more generalized over the whole body. Mild disease can resolve spontaneously, but severe forms require systemic treatment.54,55
(See Chapter 186).
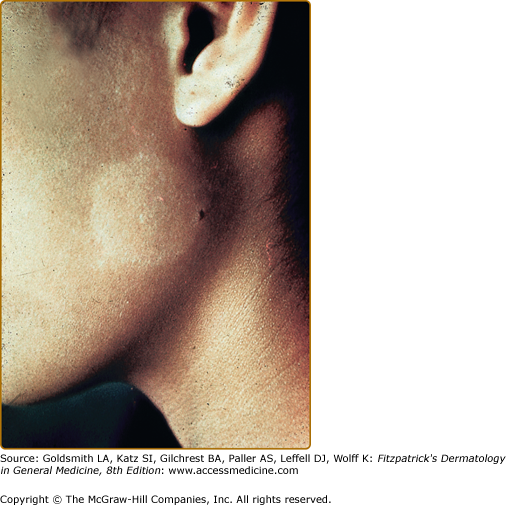
The presence of a hypopigmented lesion with reduced sensation is the hallmark of leprosy and is one of the diagnostic criteria (Fig. 75-8). Indeterminate leprosy, frequently the first manifestation of leprosy, is characterized by the presence of a few such lesions. Tuberculoid leprosy usually manifests as a limited number of well-defined, depressed, hairless lesions occur that appear hypopigmented in black patients and erythematous in white patients.56
The potential for chemicals to induce hypopigmentation was discovered in the first half of the twentieth century. Leukoderma was noticed in rubber workers exposed to the monobenzylether of hydroquinone (MBEH), a frequently used antioxidant in the rubber industry. Since then, depigmenting properties have been attributed to many chemicals and new therapeutic depigmenting agents have been developed.57
Occupational exposure to chemicals that have a destructive effect on melanocytes can result in chemical leukoderma. Agents such as para-tertiary butylphenol, para-tertiary butyl catechol, MBEH, and hydroquinone can cause permanent depigmentation. The depigmentation caused by chemicals is difficult to distinguish from idiopathic vitiligo. The former usually starts at the hands and forearms, presumptive sites of contact, but depigmentation at a distance is also possible (Fig. 75-9). Chemical leukoderma spreads by coalescence of small macules, whereas the sudden appearance of large patches with perifollicular sparing is more suggestive of vitiligo. Chemical leukoderma is very likely if several exposed workers develop depigmentation. However, not all exposed individuals develop leukoderma and it is hypothesized that the individual susceptibility is variable.57,58
Figure 75-9

Chemical leukoderma. A. O-Syl (a phenolic disinfectant)-induced chemical leukoderma that mimics vitiligo clinically. Repeated exposure is required to depigment, but antecedent clinical inflammation is not observed. B. Reversible hypomelanosis of the face in a South African woman after several weeks’ application of topical hydroquinone. Note color contrast of face to that of (untreated) hand. C. African-American factory worker depigmented from repeated exposure to monobenzyl ether of hydroquinone.
Chemicals are also employed therapeutically or cosmetically to lighten skin color. There are three main mechanisms of action for bleaching agents. Some act before melanin is synthesized by inhibiting tyrosinase transcription and glycosylation. Other agents act during melanin synthesis by inhibiting enzymes such as tyrosinase or peroxidase or act as reducing agents or radical oxygen species scavengers. Finally, some agents act after melanin synthesis because they are responsible for tyrosinase degradation, inhibition of melanosome transfer, or acceleration of skin turnover, leading to depigmentation.59
MBEH was used as a lightening agent until its ability to cause total permanent and often confetti-like depigmentation became apparent. It should be used only to produce total depigmentation in patients with diffuse vitiligo. A formulation with 20% MBEH is usually prescribed for that purpose. Hydroquinone is one of the most popular depigmenting substances and is frequently used for the treatment of melasma in concentrations between 2% and 5%. Concentrations higher than 5% incur a risk of permanent depigmentation. Adverse effects are irritation, postinflammatory pigmentation, and exogenous ochronosis.57,59–61
Heat, freezing, X-ray, ionizing radiation, UV irradiation, and laser light can cause hypopigmentation or permanent depigmentation by damaging melanocytes, leading to destruction or impaired function.62
(See Chapter 65).
Lichen sclerosus typically presents as a pruritic erythematous patch in the early stage, evolving to a depigmented atrophic plaque with porcelain white appearance.
Mechanisms believed to play a role in this idiopathic leukoderma include decreased melanin production, blocked transfer of melanosomes to keratinocytes, and loss of melanocytes.
(See Chapter 124).
Regression in a primary melanoma lesion typically causes a depigmented scar-like area within the lesion.
Melanoma-associated hypo- or depigmentation, also known as leukoderma acquisitum centrifugum, can occur around the primary melanoma or metastases or at distant sites. The latter is often called vitiligo, although the resemblance is limited. Whereas vitiligo depigmentation is usually symmetric and spreads centripetally to the trunk, melanoma-associated hypo- or depigmentation tends to be extensive, patchy, and asymmetric. The lesions may be mottled (hypomelanotic) or milk white (amelanotic). In most cases, leukoderma appears simultaneously with the finding of metastases. Histologic examination of lesions shows a decrease or complete absence of melanocytes. Macromelanocytes with stubby dendrites can also be observed. Melanoma with associated leukoderma may be associated with a better survival rate than comparably advanced lesions without epidermal pigment loss. Strong evidence suggests that melanoma-associated leukoderma results from host immune reaction against the malignancy, involving humoral and cellular mechanisms. Both passive and active immunotherapeutic strategies used in the treatment of melanoma have been associated with leukoderma (Fig. 75-10).63,64
Figure 75-10
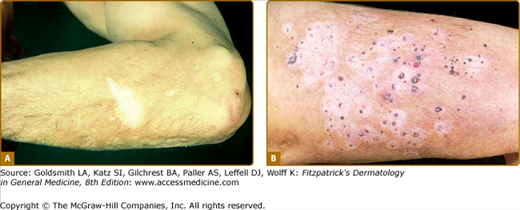
Melanoma-associated leukoderma. A. This hypomelanosis may resemble vitiligo clinically and be characterized by an absence of melanocytes. It may be associated with a favorable prognosis. B. These macules in a different patient developed after the patient developed metastases to the skin. They surround the individual metastases.
Nutritional disorders and endocrinopathies mainly cause hyperpigmentation. Diseases such as copper deficiency, vitamin B12 deficiency, kwashiorkor, Addison disease, hyperthyroidism, and diabetes can also be associated with hypopigmentation, but are discussed under Section “Hypermelanosis” because hyperpigmentation is their main feature.
(See Chapter 151).
Hypothyroidism is frequently associated with cutaneous alterations. The skin is pale due to anemia and vasoconstriction. Yellowish discoloration of the palms, soles, and nasolabial folds is caused by an accumulation of carotene in the stratum corneum. The associated hypercarotinemia results from reduced capacity of the liver to convert β-carotene to vitamin A. Vitiligo has been associated with autoimmune hypothyroidism.65–67
Panhypopituitarism results from a variety of conditions that compromise the anterior pituitary. As a consequence, release of pituitary-derived factors including melanocyte-stimulating hormone (MSH), thyroid-stimulation hormone, adrenocorticotrophic hormone, luteinizing hormone, follicle-stimulating hormone, growth hormone, and vasopressin is decreased. Due to the reduction of circulatory pituitary hormones, production of cortisol, thyroxine, estrogens, and testosterone in target organs is lowered.
Patients suffering from panhypopituitarism look pale due to anemia and decreased cutaneous blood flow. As well, generalized hypopigmentation results from decreased adrenocorticotrophic and MSH that stimulate epidermal melanogenesis.66
Castrated human males are characteristically pale and their genital skin is not hyperpigmented as in normal men. An impaired tanning response to UV radiation has been described. Administration of testosterone makes the skin turn darker and restores the tanning response.68
Loss of hair and skin pigmentation due to selenium deficiency has been described in children receiving long-term total parenteral nutrition. After selenium supplementation, skin and hair darken.69
Acquired copper deficiency occurs in severely malnourished infants. Hypopigmentation of the hair is attributed to copper deficiency, presumably because tyrosinase is a copper-dependent enzyme, but as multiple nutritional deficiencies tend to coexist pathogenesis is difficult to confirm.70
Idiopathic guttate hypomelanosis (IGH) is an acquired leukoderma, characterized by discrete, round, or oval porcelain-white macules of approximately 2–5-mm diameter, which increase in number with aging (Fig. 75-11). Any associated hairs often remain pigmented. Lesions are found in a photodistribution and tend to occur in chronically sun-damaged skin. They are often seen pretibially and on the forearms, although they may also arise on other sun-exposed areas, including the face, neck, and shoulders. IGH has been hypothesized to be UV-induced, although controversy exists. Some suggest that IGH may reflect the normal aging process.71
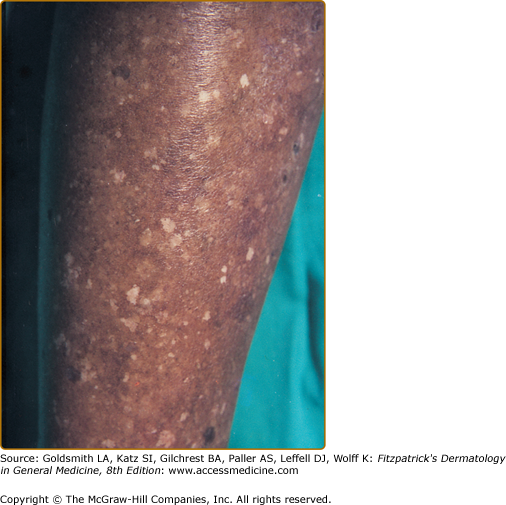
Histologically, IGH lesions are characterized by slight basket-weave hyperkeratosis with epidermal atrophy and flattening of the rete pegs. Lesions show a decrease in melanocytes and melanin content of the affected epidermis and pigment granules are irregularly distributed.
A variety of therapies with variable success are described, including cryotherapy, superficial dermabrasion, topical steroids, and topical retinoids.72,73
Leukoderma punctata was first described by the development of multiple punctiform hypopigmented and achromic spots after several months of PUVA treatment.74 Later, similar cases were described after UVB therapy for psoriasis and after topical PUVA in one case of segmental vitiligo.75
Lesions are predominantly present on the extremities, upper back and chest. They are round or oval, sharply demarcated, and small (0.5–1.5 mm), without follicular distribution. Spontaneous reduction of the leukodermic lesions has been observed.74
It has been suggested that phototoxicity damage to keratinocytes and melanocytes is the etiologic factor. Leukoderma punctatum is suggested to be distinct from IGH on the basis of the clinical and histologic features. In IGH, lesions are larger and spontaneous resolution is not reported. Ultrastructurally, leukoderma punctata demonstrates slight-to-severe damage of keratinocytes and melanocytes not reported in IGH.
Hair graying or canities is a process of chronological aging and occurs regardless of gender or race.76 The age of onset, which appears to be hereditary, is usually in the fourth decade. The average age for whites is mid-30s, for Asians late 30s, and for Africans mid-40s. Premature canities (before 20s or 30s) can be associated with pernicious anemia, hyper/hypothyroidism, osteopenia, and several rare syndromes like progeria and pangeria (Werner syndrome). Graying usually appears at the temples first, then the vertex, and, finally, the occiput. Beard and body hair are affected later. Gray hairs seem to be thicker and longer than normally pigmented hairs. The perception of “gray hair” derives in large part from the admixture of pigmented and white hair, but individual hair follicles can indeed exhibit pigment dilution or suboptimal melanocyte–cortical keratinocyte interactions during the graying process.77–79 An acute episode of alopecia areata may result in a very sudden “overnight” graying (so-called canities subita) that is caused by the preferential loss of pigmented hair in this immune-mediated disorder.
Chronic hemodialysis patients frequently show disorders of skin pigmentation, primarily involving hyperpigmentation of sun-exposed body areas. Hypopigmentation of skin and hair is rather exceptional but occurs, possibly due to a disturbance of phenylalanine metabolism.
PMH is an entity that affects the trunk with nummular, hypopigmented nonscaly macules. It affects young adults, mainly women. Although described in people of mixed racial ancestry (known as Creole dyschromia),80 it is seen in all races. It can be mistaken for PA and pityriasis versicolor. Topical and systemic antifungal treatment and topical steroids are ineffective, but the disorder may resolve, sometimes temporarily, after sun exposure or phototherapy.
The color of the skin is determined by several chromophores, usually predominantly melanin pigment, but the hemoglobin content of the skin also contributes to the skin color. The pale skin color observed in anemia is due to decreased levels of circulating oxyhemoglobin and is proportional to the severity of the anemia.
Bier spots are small, irregular apparently hypopigmented areas, usually on arms and legs in young adults, resulting in a reticulated appearance. The surrounding skin is erythematous and blanches with pressure causing the “hypopigmented” macules to disappear. The condition is a vascular anomaly with vasoconstriction in the pale areas and venodilatation in erythematous skin.81,82
(See Chapter 18).
Woronoff’s ring represents a blanched halo surrounding a psoriatic lesion. It is observed after UV treatment or topical steroid treatment, but may also occur in untreated psoriasis. The pathogenesis is not clear. A decreased melanin content has been found in both psoriatic and halo epidermis suggesting a true hypomelanosis. A local decreased prostaglandin synthesis with decreased vasodilatation or a diffusion of anti-inflammatory mediators from the psoriatic lesion to the halo is also postulated.
Cutaneous edema produces an appearance of leukoderma that is not true hypomelanosis. Decreased absorption of light, reduced capillary blood flow, and increased dermal thickness may contribute to the pale appearance of the skin.
Hypermelanosis
 The perceived color of melanin depends on the depth of the pigmentation in the skin. Melanin appears black when located in the stratum corneum and upper epidermis, brown when located in the epidermis, and gray to blue when it is located in the deeper dermis.
The perceived color of melanin depends on the depth of the pigmentation in the skin. Melanin appears black when located in the stratum corneum and upper epidermis, brown when located in the epidermis, and gray to blue when it is located in the deeper dermis.
 The color blue occurs when there is melanin localized within the deeper parts of the skin because the portions of visible light with shorter wavelengths (blue–violet end of spectrum) are more dispersed than portions with longer wavelengths (red end of visible spectrum). The tendency of blue light to be scattered when it travels through a turbid medium is also known as the Tyndall effect.83,84
The color blue occurs when there is melanin localized within the deeper parts of the skin because the portions of visible light with shorter wavelengths (blue–violet end of spectrum) are more dispersed than portions with longer wavelengths (red end of visible spectrum). The tendency of blue light to be scattered when it travels through a turbid medium is also known as the Tyndall effect.83,84
Descriptions of the rare and poorly understood conditions, arterial dissection with lentigines, and progressive familial hyperpigmentation are provided within.
 Arterial dissection with lentiginosis was reported by Schievink et al in 1995.85 They described two patients and their families, with a history of arterial dissection and the progressive development of numerous lentigines first noted in childhood and increasing in number until early adulthood. Some lesions faded or disappeared with increasing age. They consider this syndrome to be genetic in origin and suggest autosomal recessive inheritance. The histologically confirmed lentigines were of brown-to-black pigmentation, approximately 2–4 mm in diameter. They were present on sun-exposed and sun-protected areas on the trunk and particularly on the extremities, including palms, soles, fingers, and toes.
Arterial dissection with lentiginosis was reported by Schievink et al in 1995.85 They described two patients and their families, with a history of arterial dissection and the progressive development of numerous lentigines first noted in childhood and increasing in number until early adulthood. Some lesions faded or disappeared with increasing age. They consider this syndrome to be genetic in origin and suggest autosomal recessive inheritance. The histologically confirmed lentigines were of brown-to-black pigmentation, approximately 2–4 mm in diameter. They were present on sun-exposed and sun-protected areas on the trunk and particularly on the extremities, including palms, soles, fingers, and toes.
 Several families with a diverse spectrum of progressive hyperpigmentation have been described,86 thought to be inherited as an autosomal dominant. Recently, progressive familial hyperpigmentation was reported to be caused by a KIT ligand (KITLGN365) mutation, with a gain-of-function effect on melanin synthesis.87
Several families with a diverse spectrum of progressive hyperpigmentation have been described,86 thought to be inherited as an autosomal dominant. Recently, progressive familial hyperpigmentation was reported to be caused by a KIT ligand (KITLGN365) mutation, with a gain-of-function effect on melanin synthesis.87
 The disorder is characterized by gradually increasing skin pigmentation starting at birth or in early infancy, without systemic anomalies. Pigmentation is variable, described as a diffuse and blotchy hyperpigmentation (various shades of brown, black, or bronze), occasionally in combination with hypopigmentated macules and/or numerous lentigines.88 Mucous membranes and the cornea can be affected as well. With aging the pigmentation can fade, resulting in a bronze or mottled appearance.
The disorder is characterized by gradually increasing skin pigmentation starting at birth or in early infancy, without systemic anomalies. Pigmentation is variable, described as a diffuse and blotchy hyperpigmentation (various shades of brown, black, or bronze), occasionally in combination with hypopigmentated macules and/or numerous lentigines.88 Mucous membranes and the cornea can be affected as well. With aging the pigmentation can fade, resulting in a bronze or mottled appearance.
 Histology shows markedly increased amounts of melanin in the epidermis, particularly in the basal layer. The number and size of melanocytes is normal and only rare melanophages are observed in the dermis.
Histology shows markedly increased amounts of melanin in the epidermis, particularly in the basal layer. The number and size of melanocytes is normal and only rare melanophages are observed in the dermis.
Linear and whorled nevoid hypermelanosis (LWNH) is characterized by hyperpigmented macules in streaky configuration along the lines of Blaschko without preceding inflammation or atrophy.89 Similar cases have been described under different descriptive names (zosteriform hyperpigmentation, zosteriform lentiginous nevus, zebra-like hyperpigmentation). Lesions are typically located on the trunk and limbs and do not cross the midline. Face, palms, soles, eyes, and mucous membranes are spared. Kalter et al described LWNH as having onset within a few weeks of age and progression during the initial years of life.89 The pigmentation can fade gradually with increasing age. Several cases have been reported with both hyper- and hypopigmentations. Pigmentary mosaicism is a useful term to encompass all these different phenotypes.90
The presence of mosaicism has been confirmed in a few cases (mosaic trisomy 7, 14, 18, 20, and X-chromosomal mosaicism) by chromosomal analysis on lymphatic cultures or dermal fibroblasts.91
LWNH may be differentiated from incontinentia pigmenti (IP) and epidermal nevus.89
Extracutaneous abnormalities have been observed in a number of LWNH cases, including developmental and growth retardation, facial and body asymmetry, ventricular septal defect, and pseudohermaphroditism.
Histologic examination reveals increased pigmentation of the basal layer and prominence or vacuolization of melanocytes. Pigment incontinence is usually but not always absent.92
IP, also known as Bloch–Sulzberger syndrome, was first described by Garrod et al in 1906. It is an X-linked, dominantly inherited disorder, reported primarily in females, and believed to be embryonic lethal in the majority of males. In most cases, IP is due to a mutation in the gene NEMO [nuclear factor κB (NF-κB) essential modulator] on the X chromosome at Xq28.93–96 In IP females, inactivation of one of the two X chromosomes through a process termed lyonization occurs during embryogenesis. Epidermal cells expressing the defective NEMO gene give rise to typical skin lesions along the lines of Blaschko, reflecting the embryonic migration path of the affected keratinocytes. Lesions usually proceed through four cutaneous stages, sometimes with some overlap: (1) vesicular stage (from birth or shortly thereafter), (2) verrucous stage (between 2 and 8 weeks of age), (3) hyperpigmented stage (several months of age into adulthood), followed by (4) hypopigmentation stage (from infancy through adulthood) (Fig. 75-12). A significant percentage of IP patients have ocular, dental, skeletal, and CNS anomalies.97
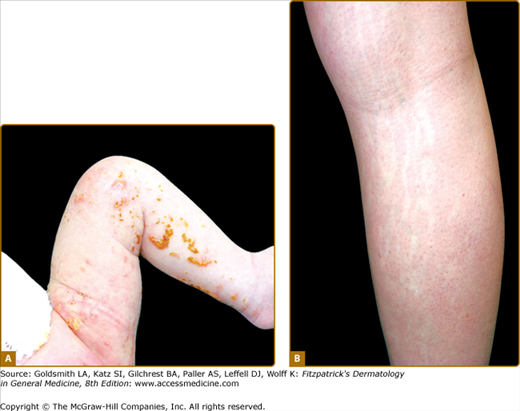
The cutaneous lesions in the first stage represent the population of NEMO-deficient cells that fail to activate NF-κB, leading to apoptosis, as NF-κB normally protects against tumor necrosis factor-induced apoptosis. The number of NEMO-deficient cells decreases secondary to apoptosis and is replaced by cells expressing the normal allele. Subsequently, the inflammatory and vesicular stage ends. The hyperproliferation in the second stage is likely due to compensatory proliferation of normal NEMO keratinocytes. Hyperpigmentation in the third stage results from incontinence of melanin pigment from the destroyed epidermis into the dermis.
The hyperpigmentation appears in streaks and whorls along the lines of Blaschko and is usually most pronounced on the trunk, but can also appear on the extremities. The degree of hyperpigmentation varies among individuals. Histologically, the areas of pigmentation show many melanin-laden melanophages, extensive deposits of melanin in the basal cell layer and dermis. There is vacuolization and degeneration in the epidermal basal cell layer. Usually, the hyperpigmentation fades gradually after several years and the skin can become hypopigmented (stage 4), which represents postinflammatory dermal scarring. The hypopigmentation stage is characterized by linear, atrophic, hairless scars following the Blaschko’s lines.
Histologically, the number of melanocytes seems to be normal, although a reduced number of melanocytes also has been reported. The epidermis is thinner and there is an absence or reduction of skin appendages in the dermis that may contribute to the impression of hypopigmentation.98
A beneficial effect of topical steroids and topical tacrolimus in the vesicular stage has been described.99,100
Dyskeratosis congenital (DKC) or Zinnser–Engmann–Cole syndrome is characterized by reticulate skin pigmentation, nail atrophy, leukoplakia, and bone marrow failure. Bone marrow failure and malignancy develop in the second and third decades. In all characterized cases of DKC, the causative mutations are present in components of the telomerase complex. Rapidly dividing somatic cells express low but detectable levels of telomerase activity that slows the otherwise progressive telomere shortening that occurs with each cycle of DNA replication and eventually leads to cellular senescence (permanent loss of proliferative capacity). It is now thought that DKC is due to defective telomere maintenance, limiting the proliferative capacity of hematopoietic and epithelial cells. Increased melanin synthesis is now recognized to occur in senescent melanocytes, likely accounting for the pigmentary phenotype of DKC; and critically short telomeres may force cells into “replicative crisis,” at which time activation of an alternative “ALT” mechanism for lengthening telomeres in the absence of telomerase may lead to development of malignancies. The finding of shortened telomeres in DKC was indeed the first evidence of the role of telomeres in cell biology (cellular aging).101
X-linked DKC is caused by mutations in the DKC1 gene located at Xq28, encoding for dyskerin. Females carrying one mutated allele are protected by expression of normal telomerase on the unaffected allele. In autosomal dominant DKC the majority of cases are due to mutations in TERC, the RNA component of the telomerase complex. TERT (telomerase reverse transcriptase) is affected much less often in autosomal dominant DKC. The autosomal dominant form has the better prognosis, presumably because some telomerase activity is preserved, due to the presence of an unaffected allele.
In the autosomal recessive form of DKC, mutations in telomerase-associated proteins such as NOP10, NHP2, and TINF2 are involved.102–104 Skin biopsy of hyperpigmented skin shows nonspecific changes, including epidermal atrophy, a chronic inflammatory infiltrate with numerous melanophages in the upper dermis. DKC may be confused with Fanconi’s syndrome, characterized by short stature, hypoplastic or aplastic thumbs, and a reduced number of carpal bones. Here, there is a patchy hyperpigmentation of the trunk, neck, groin, and axillary region that manifests itself earlier than in DKC, i.e., in the first few years of life.
For a discussion of Naegeli–Franceschetti–Jadassohn syndrome, dermatopathia pigmentosa reticularis, Dowling–Degos disease, Galli–Galli disease, Kitamura reticular acropigmentation, Haber’s syndrome, and Partington syndrome, see below.









