Key Words
hypersensitivity syndrome, Sweet syndrome, Schamberg disease, Stevens–Johnson syndrome, toxic epidermal necrosis, erythema, dermatosis, Churg–Strauss syndrome, Wegener granulomatosis, polyarteritis nodosa, hypersensitivity vasculitis, Henoch–Schönlein purpura, erythema elevatum diutinum, pyoderma gangrenosum
Hypersensitivity Syndromes
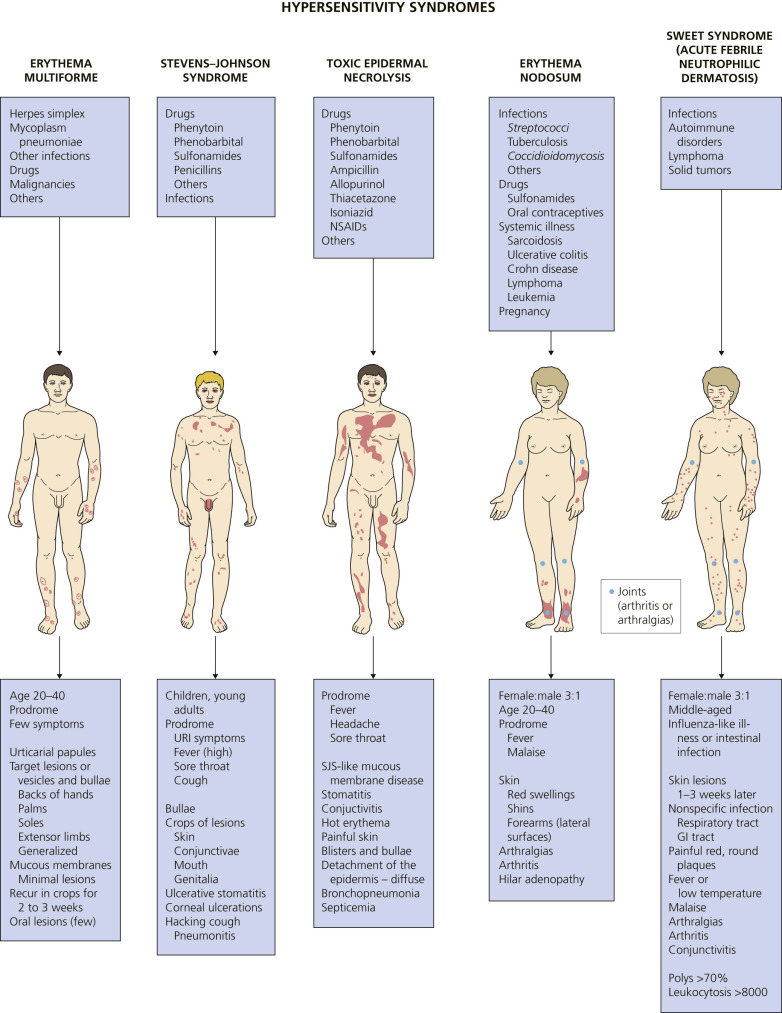
Hypersensitivity syndromes are displayed in Fig. 18.1 .
Erythema Multiforme
Erythema multiforme (EM) is a relatively common, acute, often recurrent inflammatory disease. Many factors have been implicated in the etiology of EM, including numerous infectious agents, drugs, physical agents, X-ray therapy, pregnancy, and internal malignancies ( Box 18.1 ). In approximately 50% of cases no cause can be found. EM is commonly associated with a preceding acute upper respiratory tract infection, herpes simplex virus (HSV) infection, or Mycoplasma pneumoniae infection such as primary atypical pneumonia.
Etiology
Infections (viral, bacterial, fungal, protozoal)
Viral: Herpes simplex virus 1 and 2 (most common cause), parapoxvirus, hepatitis C, Coxsackie virus, Epstein–Barr Virus, cytomegalovirus, varicella–zoster virus, adenovirus, parvovirus B-19, human immunodeficiency virus
Bacterial: Mycoplasma pneumonia (especially children), Chlamydophila psittaci , Salmonella , mycobacterial tuberculosis
Fungal infections: Histoplasma capsulatum , dermatophytes
Medications (oxicam drugs, allopurinol, phenobarbital, phenytoin, valproic acid, sulfonamides, penicillins, erythromycin, nitrofurantoin, tetracyclines, chlormezanone, acetylsalicylic acid, statin medications (simvastatin and pravastatin [with sun exposure]), and TNF-α inhibitors (adalimumab, infliximab, etanercept), imiquimod, barbiturates
Vaccinations (diphtheria–tetanus–pertussis), cancer immunotherapy (alectinib, nivolumab, vemurafenib)
Endogenous and exogenous sexual hormones
Taurine-containing energy drinks.
Allergic contact dermatitis (Rhus, nickel)
Management
Acute Erythema Multiforme
Observation
Oral antihistamines
Topical steroids
Acyclovir
Prednisone
Recurrent Erythema Multiforme
Oral acyclovir
Valacyclovir
Famciclovir
Dapsone
Hydroxychloroquine
Azathioprine
Cyclosporine
Thalidomide
TNF, tumor necrosis factor.
Classification.
A classification system, based on the pattern and distribution of cutaneous lesions, separates erythema multiforme major from Stevens–Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) (see the section titled Stevens–Johnson Syndrome ). Erythema multiforme differs from SJS and TEN by occurrence in younger males, frequent recurrences, less fever, milder mucosal lesions, and lack of association with collagen vascular diseases, human immunodeficiency virus (HIV) infection, or cancer. Recent or recurrent herpes is the principal risk factor for EM. Compared to EM, drugs have higher etiologic fractions for SJS and TEN.
Recurrent EM.
Herpes-associated EM develops in only a few of the many individuals who experience recurrent HSV infection. EM develops in some adults and children after each episode of herpes simplex. Other reported causes are complex aphthosis, infection other than HSV infection, Mycoplasma pneumoniae; infection with hepatitis C virus; recurrent vulvovaginal candidiasis; or recurrent infections related to previous interventions for Crohn disease (i.e., bacteremia, line infections [intravenous or central lines], and cutaneous infections). A medication (acetaminophen) induced recurrent EM in one patient. In another patient, menses induced monthly episodes of recurrent EM, which occurred before the onset of menses. More than half of patients do not have an identifiable cause for recurrent EM.
Pathogenesis
Studies suggest that immune complex formation and subsequent deposition in the cutaneous microvasculature may play a role in the pathogenesis of EM. Circulating complexes and deposition of C3, immunoglobulin M (IgM), and fibrin around the upper dermal blood vessels have been found in the majority of patients with EM. Erythema multiforme has a high-density cell infiltrate rich in T lymphocytes. In contrast, TEN is characterized by a cell-poor infiltrate in which macrophages and dendrocytes predominate.
Clinical Manifestations
The prodromal symptoms, morphologic configuration of the lesions, and intensity of systemic symptoms vary. Milder forms of the disease may be preceded by malaise, fever, or itching and burning at the site where the eruption will occur. The cutaneous eruptions are most distinctive, and classification is based on their form. Mucosal lesions may occur in up to 70% of cases. The most common sites are the lips and buccal mucosa. The differential diagnosis is listed in Box 18.2 .
Bullous pemphigoid
Dermatitis herpetiformis
Drug eruptions
Leukocytoclastic vasculitis
Lupus erythematosus
Pityriasis rosea
Polymorphic light eruption
Stevens–Johnson syndrome
Toxic epidermal necrolysis
Urticaria
Urticarial vasculitis
Viral exanthems
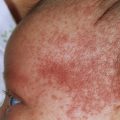
Target Lesions and Papules.
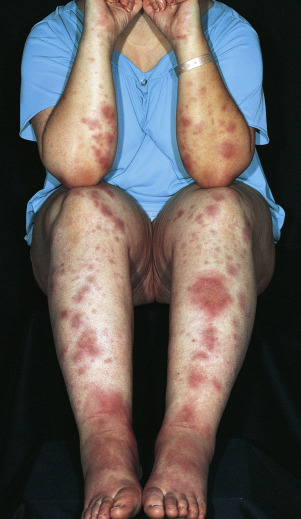
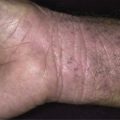
Target lesions and papules are the most characteristic eruptions. Dusky red, round maculopapules appear suddenly in a symmetric pattern on the backs of the hands and feet and on the extensor aspect of the forearms and legs. The trunk may be involved in more severe cases. Early lesions itch, burn, or are asymptomatic. The diagnosis may not be suspected until the nonspecific early lesions evolve into target lesions during a 24 to 48 hour period ( Figs. 18.2 to 18.4 ). The classic “iris” or target lesion results from centrifugal spread of the red maculopapule to a circumference of 1 to 3 cm as the center becomes cyanotic, purpuric, or vesicular. The mature target lesion consists of two distinct zones: an inner zone of acute epidermal injury with necrosis or blisters and an outer zone of erythema. There may be a middle zone of pale edema. Partially formed targets with annular borders or target lesions on the palms and soles are less distinctive and clinically resemble urticaria . Individual lesions heal in 1 or 2 weeks without scarring but with hypopigmentation or hyperpigmentation, while new lesions appear in crops.
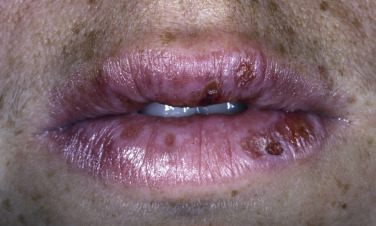
Bullae and erosions may be present in the oral cavity. The entire episode lasts for approximately 1 month.
Laboratory Investigations.
Elevated erythrocyte sedimentation rate (ESR) and moderate leukocytosis are found in the more severe cases. Biopsy is performed for atypical cases. Direct immunofluorescence may be needed to exclude other bullous diseases.
Treatment.
Mild cases are not treated. Patients with many target lesions respond rapidly to a 1- to 3-week course of prednisone. Prednisone (40 to 80 mg/day) is continued until control is achieved and is then tapered rapidly in 1 week. Treatment with prednisone can successfully abort a recurrence. In patients with suspected medication-induced EM, the suspected medication should be discontinued and when possible the medication should be switched to a chemically dissimilar class, to prevent cross-reactivity.
Recurrent Erythema Multiforme.
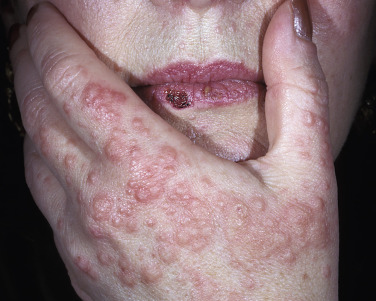
Oral acyclovir (400 mg twice a day) used continually prevents herpes-associated recurrent EM in many cases ( Fig. 18.5 ). Recurrent erythema multiforme patients should receive oral acyclovir (where HSV is not an obvious precipitating factor) for 6 months. Valacyclovir and famciclovir are absorbed better than acyclovir and may be used for patients who do not respond to acyclovir. If these treatments fail, dapsone or antimalarial drugs may be tried. Dapsone (100 to 150 mg daily) may induce partial or complete remission. Azathioprine may be successful in patients with severe disease for whom all other treatments have failed. The response to treatment is dose-dependent (100 to 150 mg daily). The condition recurred on discontinuation of therapy. Mycophenolate mofetil provided partial or complete response in six of eight patients. Intramuscularly administered human immunoglobulin, cyclosporine, thalidomide, interferon-α (in cases secondary to hepatitis C virus infection), and cimetidine are effective in select cases. More recently, tofacitinib and apremilast have successfully treated recurrent EM.
Stevens–Johnson Syndrome/Toxic Epidermal Necrolysis Spectrum of Disease
Stevens–Johnson syndrome (SJS) and TEN are rare diseases with an annual incidence of 1.2 to 6 and 0.4 to 1.2 per million persons, respectively. Studies have shown that SJS and TEN result from the cumulative effect of risk factors such as the chemical structure of the drug, host genetic factors such as human leukocyte antigen alleles (HLA type), drug metabolism and T lymphocyte clones. When these factors align perfectly, then SJS and TEN may result. Table 18.1 shows the genetic associations of SJS and TEN in select populations and Table 18.2 shows high-risk drugs and their relationship to select patient populations. Patients at greatest risk are those with slow acetylator genotypes, immunocompromised patients (e.g., HIV infection, lymphoma), and patients with brain tumors who are undergoing radiotherapy and receiving antiepileptics. Drugs are implicated in more than 95% of patients with TEN. The etiology of SJS is less well-defined because about 50% of reported SJS cases are claimed to be drug related. The most frequently implicated drugs are shown in Box 18.3 .
| Drug Classification | Culprit Drug | SJS and/or TEN | HLA Allele and Cyp | Ethnicity and References |
|---|---|---|---|---|
| Antibiotics | Sulfonamide | TEN | A *29, B *12, DR *7 | European |
| Sulfamethoxazole | SJS/TEN | B *38 | European | |
| Anticonvulsants | Carbamazepine | SJS/TEN | B *15 : 02 | Han Chinese, Thai, Indian, Malaysian |
| SJS/TEN | B *15 : 11 | Japanese, Korean, Han Chinese | ||
| SJS/TEN | B *59 : 01 | Japanese | ||
| SJS/TEN | A *31 : 01 | Japanese, northern European | ||
| Lamotrigine | SJS/TEN | B *15 : 02 | Han Chinese | |
| Oxcarbazepine | SJS/TEN | B *15 : 02 | Han Chinese | |
| Phenytoin | SJS/TEN | B *15 : 02 | Han Chinese, Thai | |
| SJS/TEN | CYP2C9 *3 | Han Chinese, Japanese, Malaysian | ||
| Antiglaucoma drugs | Methazolamide | SJS/TEN | B *59 : 01, CW *01 : 02 | Korean and Japanese |
| Antiretrovirals | Nevirapine | SJS/TEN | CYP2B6 | African in Mozambique |
| C *04 : 01 | African in Malawi | |||
| NSAIDs | Oxicam | SJS/TEN | A *2, B *12 | European |
| TEN | B *73 | |||
| Xanthine oxidase inhibitors | Allopurinol | SJS/TEN | B *58 : 01 | Han Chinese, Thai, Japanese, Korean, European |
| Drugs | General Population , a | Children , b | Africa , c |
|---|---|---|---|
| Allopurinol | [✓] Highest incidence in Europe and Israel | [✓] | |
| Antibacterial sulfonamides | [✓] | [✓] | [✓] Highest incidence in Africa |
| Antiepileptic agents | [✓] Carbamazepine Lamotrigine Phenobarbital Phenytoin | [✓] Carbamazepine Lamotrigine Phenobarbital | [✓] |
| NSAIDs | [✓] Oxicam NSAIDs | [✓] | |
| Nevirapine | [✓] | [✓] | |
| Sulfasalazine | [✓] | ||
| Antituberculosis agents | [✓] | ||
| Aminopenicillin | [✓] | ||
| Analgesics | [✓] |
a Information collected from population in Europe and Israel as part of the EuroSCAR study.
b Information collected from population in Europe and Israel as part of the SCAR and EuroSCAR studies.
Prescribing High-Risk Drugs: Practical Recommendations
A few medications are associated with high risks of SJS or TEN. Prescribing one of them requires thorough evaluation of expected benefits.
- •
Nevirapine
- •
Lamotrigine
- •
Carbamazepine
- •
Phenytoin
- •
Phenobarbital
- •
Co-trimoxazole and other antiinfective sulfonamides
- •
Sulfasalazine
- •
Allopurinol
- •
Oxicam NSAIDs
A delay of 4 to 28 days between beginning of drug use and onset of the adverse reaction is the most suggestive timing supporting drug causality in SJS or TEN.
In cases of exposure to several medications with high expected benefits, the timing of administration is important to determine which one(s) must be stopped and if some may be continued or reintroduced.
The risks of various antibiotics to induce SJS and TEN are within the same order of magnitude, but substantially lower than the risk of antiinfective sulfonamides.
- •
Valproic acid does not seem to be a major risk factor by itself.
- •
Sulfonamide-related diuretics and antidiabetics do not appear to be risk factors.
NSAID, nonsteroidal antiinflammatory drug; SJS, Stevens–Johnson syndrome; TEN, toxic epidermal necrolysis.
Stevens–Johnson syndrome and TEN have traditionally been considered the most severe forms of EM. It was proposed that EM major is distinct from SJS and TEN on the basis of clinical criteria. The proposed concept is to separate an EM spectrum from an SJS/TEN spectrum. EM, characterized by typical target lesions, is a postinfectious disorder, often recurrent but with low morbidity. The second spectrum (SJS/TEN), characterized by widespread blisters and purpuric macules, is usually a severe drug-induced reaction with high morbidity and a poor prognosis. In this concept, SJS and TEN might be only types of the same drug-induced process that vary in severity.
A three-grade classification has been proposed and the degree of cutaneous involvement in TEN and SJS is shown in Box 18.4 .
Grade 1: SJS mucosal erosions and epidermal detachment less than 10%
Grade 2: Overlap SJS/TEN epidermal detachment between 10% and 30%
Grade 3: TEN epidermal detachment more than 30%
Protocols have been developed for SJS and TEN ( Table 18.3 ). The algorithm of drug causality for epidermal necrolysis (ALDEN) has been shown to predict if a drug caused SJS and TEN ( Table 18.4 ).
| 5Ds | Key Protocol Elements |
|---|---|
| D iagnosis | Diagnosis is based on cutaneous and mucous membrane manifestations, systemic involvement, and histologic findings Tools for evaluation
|
| D rug exposure (timing) | All medications must be considered, especially new drugs taken in the 8 weeks prior to the skin reaction; drug exposure analysis by a timeline chart of the patient’s illness Tools for evaluation
|
| D ifferential diagnosis |
|
| D etermine probabilities of causality |
|
| D etermine severity | Hemodynamic status and systemic involvement SCORTEN score
|
| Histologic findings: extent of dermal mononuclear inflammation and epidermal necrosis |
| Criterion | Values | Rules to Apply | |
|---|---|---|---|
| Delay from initial drug component intake to onset of reaction (index day) | Suggestive +3 Compatible +2 Likely +1 Unlikely −1 Excluded −3 | From 5 to 28 days From 29 to 56 days From 1 to 4 days >56 Days Drug started on or after the index day | −3 to 3 |
| In case of previous reaction to the same drug, only changes for: Suggestive: +3: from 1 to 4 days Likely: +1: from 5 to 56 days | |||
| Drug present in the body on index day | Definite 0 | Drug continued up to index day or stopped at a time point less than five times the elimination half-life a before the index day | −3 to 0 |
| Doubtful −1 | Drug stopped at a time point prior to the index day by more than five times the elimination half-life a but liver or kidney function alterations or suspected drug interactions b are present | ||
| Excluded −3 | Drug stopped at a time point prior to thde index day by more than five times the elimination half-life, a without liver or kidney function alterations or suspected drug interactions b | ||
| Prechallenge/rechallenge | Positive specific for disease and drug: 4 | SJS/TEN after use of same drug | −2 to 4 |
| Positive specific for disease or drug: 2 | SJS/TEN after use of similar c drug or other reaction with same drug | ||
| Positive unspecific: 1 | Other reaction after use of similar c drug | ||
| Not done/unknown: 0 | No known previous exposure to this drug | ||
| Negative −2 | Exposure to this drug without any reaction (before or after reaction) | ||
| Dechallenge | Neutral 0 | Drug stopped (or unknown) | −2 or 0 |
| Negative −2 | Drug continued without harm | ||
| Type of drug (notoriety) | Strongly associated 3 | Drug of the “high-risk” list according to previous case–control studies | 1 to 3 |
| Associated 2 | Drug with definite but lower risk according to previous case–control studies | ||
| Suspected 1 | Several previous reports, ambiguous epidemiology results (drug “under surveillance”) | ||
| Unknown 0 | All other drugs including newly released ones | ||
| Not suspected −1 | No evidence of association from previous epidemiology study with sufficient number of exposed controls c | ||
| Intermediate score = total of all previous criteria | −11 to 10 | ||
| Other cause | Possible −1 | Rank all drugs from highest to lowest intermediate score | −1 |
| If at least one has an intermediate score >3, subtract 1 point from the score of each of the other drugs taken by the patient (another cause is more likely) | |||
| Final score −12 to 10 | |||
a Drug (or active metabolite) elimination half-life from serum and/or tissues (according to pharmacology textbooks), taking into account kidney function for drugs predominantly cleared by kidney and liver function for those with high hepatic clearance.
b Suspected interaction was considered when more than five drugs were present in a patient’s body at the same time.
c Similar drug = same ATC code up to the fourth level (chemical subgroups).
Predicting Patient Outcome.
A scoring system for TEN named the SCORTEN severity of illness score was developed to predict patient mortality ( Table 18.5 ). Seven parameters are given one point if positive and zero if negative. Computing the sum of the scores results in a “SCORTEN” ranging from 0 to 7, with a score of 0 or 1 predicting a mortality of 3.2%, and a score of 5 or greater predicting a mortality of greater than 90%.
| SCORTEN VARIABLES * | ||
| Extent of epidermal detachment >10% | SCORTEN Total | Predicted Death Rate (%) |
| Age >40 years | 0–1 | 3.2 |
| History of malignancy | 2 | 12.1 |
| Heart rate >120 bpm | 3 | 35.3 |
| Urea >10 mmol/L | 4 | 58.3 |
| Glucose >14 mmol/L | ≥5 | 90 |
| Bicarbonate <20 mmol/L | ||
* One point is scored for each variable in the left column present in the first 24 hours after admission.
Evolution of Lesions in Stevens–Johnson Syndrome and Toxic Epidermal Necrolysis.
Skin lesions appear on the trunk first and then spread to the neck, face, and proximal upper extremities. The distal portions of the arms are usually spared, but the palms and soles can be an early site of involvement. Erythema and erosions of the buccal, ocular, and genital mucosa are present in more than 90% of patients. The epithelium of the respiratory tract is involved in 25% of cases of TEN, and gastrointestinal (GI) lesions can also occur. Skin lesions are tender, and mucosal erosions are painful. First, lesions appear as dusky-red or purpuric macules that have a tendency to coalesce. The macular lesions assume a gray hue. This process can occur in hours or take several days. The necrotic epidermis then detaches from the dermis and fluid fills the space to form blisters. The blisters break easily (flaccid) and extend sideways with slight thumb pressure as more necrotic epidermis is displaced laterally (Nikolsky sign). The skin looks like wet cigarette paper as it is slid away by pressure to reveal large areas of bleeding dermis.
Stevens–Johnson Syndrome
Vesiculobullous disease of the skin, mouth, eyes, and genitals is called Stevens–Johnson syndrome. The disease occurs most often in children and young adults. The cutaneous eruption is preceded by symptoms of an upper respiratory tract infection. A harsh, hacking cough and patchy changes on chest radiograph examination indicate pulmonary involvement. Patients with limited disease may be weak and lethargic, but the prognosis is good with conservative treatment. Mortality approaches 10% for patients with extensive disease. Fever is high during the active stages. Oral lesions may continue for months.
Initial Symptoms.
Initial symptoms are fever, stinging eyes, and pain with swallowing. They precede cutaneous manifestations by 1 to 3 days.
Skin Lesions.
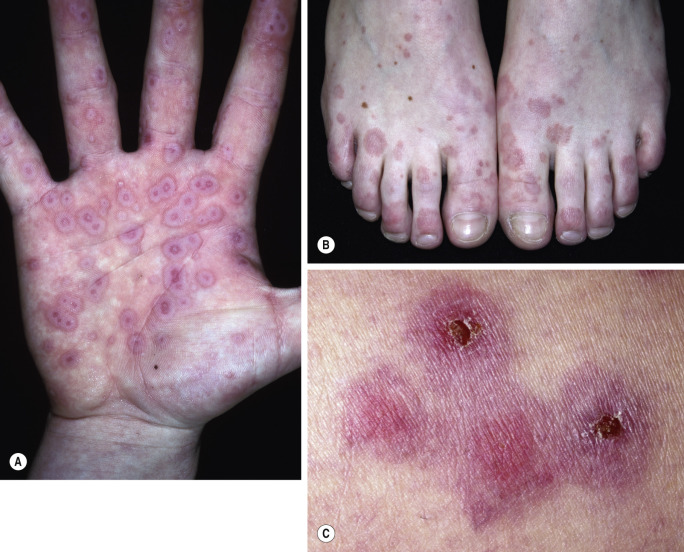
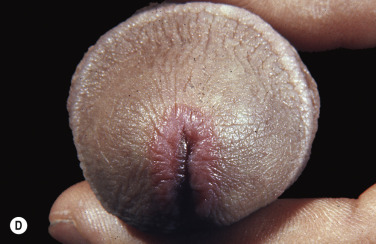
Skin lesions in SJS are flat, atypical targets; or purpuric maculae that are widespread or distributed on the trunk first and then spread to the neck, face, and proximal upper extremities. The palms and soles may be an early site of involvement ( Figs. 18.6 and 18.7 ). This is in contrast to the lesions in erythema multiforme, which consist of typical or raised atypical targets or raised edematous papules that are located on the extremities and/or the face. New crops of lesions appear, but the disease is self-limited and resolves in approximately 1 month if there are no complications.
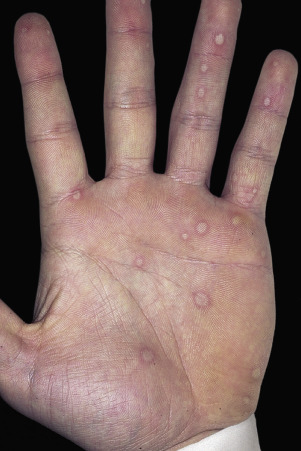
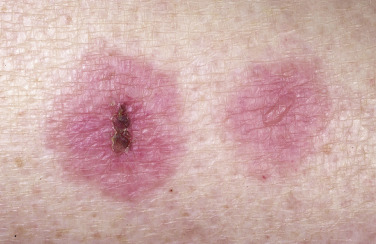
Mucosal Lesions.
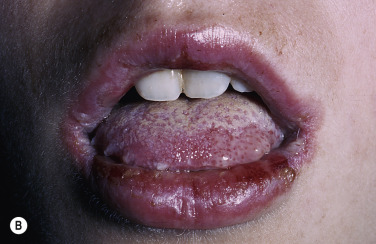
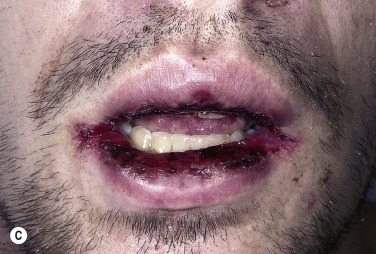
Bullae occur suddenly 1 to 14 days after the prodromal symptoms, appearing on the conjunctivae and mucous membranes of the nares, mouth ( Fig. 18.8 ), anorectal junction, vulvovaginal region, and urethral meatus. Ulcerative stomatitis leading to hemorrhagic crusting is the most characteristic feature.
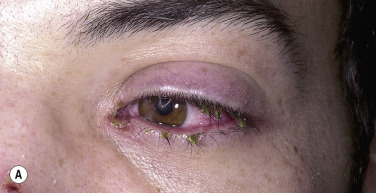
Ocular Symptoms.
Corneal ulcerations may lead to blindness. Severe ocular mucosal injury that occurs in SJS may be a precipitating factor in the development of ocular cicatricial pemphigoid, a chronic, scarring inflammation of the ocular mucosae that can lead to blindness. The time between the onset of SJS and cicatricial pemphigoid ranges from a few months to 31 years.
Etiology.
Drugs are the most common cause (see Box 18.3 ). The disease occurs most often in patients treated for seizure disorders. Upper respiratory tract infection, Mycoplasma pneumoniae infection, GI disorders, and HSV infection are all implicated. Possible causes should be pursued diligently so that recurrences can be avoided.
Diagnosis.
A skin biopsy should be performed if the classic lesions are not present. Direct immunofluorescence may be helpful in nontypical cases.
Treatment.
The use of corticosteroids remains controversial. A study of children suggests that treatment with systemic corticosteroids may be associated with delayed recovery and significant side effects. Other studies conclude that corticosteroids are beneficial and may be life-saving. Many physicians presented with a sick child who has extensive cutaneous, ocular, and oral lesions elect to treat with oral steroids; most often prednisone (20 to 30 mg twice a day) is given until new lesions no longer appear; it is then tapered rapidly.
Itching can be controlled with antihistamines. Cutaneous blisters are treated with cool, wet Burow’s compresses. Topical steroids should not be applied to eroded areas. Papules and plaques may respond to group II to V topical steroids. Frequent rinsing with lidocaine hydrochloride (Xylocaine Viscous) may relieve oral symptoms. Patients may tolerate only a liquid or soft diet. Ocular involvement is monitored by an ophthalmologist to minimize conjunctival scarring. Antiseptic eye drops and separation on synechiae are required. Vitamin A administered topically and systemically was reported to be effective for lacrimal hyposecretion. Secondary infection is treated with oral antibiotics. SJS associated with HSV may be prevented by early use of acyclovir and prednisone.
Toxic Epidermal Necrolysis
Toxic epidermal necrolysis (TEN) is a severe drug reaction involving keratinocyte death. TEN is initially seen with SJS-like mucous membrane disease; progresses to diffuse, generalized detachment of the epidermis through the dermoepidermal junction; and results in the formation of bullae and epidermal sloughing. This full-thickness loss of the epidermis results in a high death rate. Fluid loss is not a major problem; death is usually caused by overwhelming sepsis originating in denuded skin or lungs. TEN is rare, occurring in 1.3 cases per million persons per year. The death rate is 1% to 5% for SJS, and 34% to 40% for TEN. Mortality is not affected by the type of drug responsible. There is a high prevalence of HIV infection among patients with TEN. This high rate of HIV infection is linked to an increased use of sulfonamides – mainly sulfadiazine – in these patients. TEN may occur after bone marrow transplantation. It seems to be related to a drug reaction to sulfonamides as often as to acute graft-versus-host disease.
Toxic Epidermal Necrolysis Versus Staphylococcal Scalded Skin Syndrome.
This life-threatening disease is similar in appearance to staphylococcal scalded skin syndrome (SSSS), which is induced by a staphylococcal toxin. The split in SSSS, however, is high in the epidermis, just below the stratum corneum, permitting rapid healing of the epidermis without danger of infection. The diagnosis of either TEN or SSSS can be made rapidly by examination of a skin biopsy by frozen section technique.
Pathology and Pathogenesis.
Histologically there is an early mild interface dermatitis that evolves into full-thickness necrosis of the epidermis. Keratinocytes from patients with TEN were found to undergo extensive apoptosis. There is subepidermal blister formation, keratinocyte necrosis, and a sparse lymphohistiocytic infiltrate around superficial dermal blood vessels. Lymphopenia is frequently documented. Cytotoxic T cells may contribute to the pathogenesis of blister formation by causing degeneration and necrosis of drug-altered keratinocytes.
Etiology.
The causes of TEN are the same as those for SJS, but drugs are most frequently implicated in TEN. The reaction is independent of dosage. Culprit drugs include antibiotics (40%), anticonvulsants (11%), and analgesics (5% to 23%). Developing countries have a higher incidence of reactions to antituberculous drugs. The most frequent underlying diseases justifying drug treatment are infections (52.7%) and pain (36%). Challenge tests are absolutely contraindicated in SJS and TEN. TEN and other severe cutaneous adverse drug reactions may be linked to an inherited defect in the detoxification of drug metabolites. In a few predisposed patients, a drug metabolite may bind to proteins in the epidermis and trigger an immune response, leading to immunoallergic cutaneous adverse drug reaction.
Predrug Testing and Genetic Associations.
Clinical studies have demonstrated associations between some HLA genotypes and drug-induced adverse skin reactions. Allopurinol hypersensitivity syndrome (AHS) is a spectrum of reactions that includes SJS and TEN, and also systemic disease with eosinophilia, vasculitis, rash, and major end-organ disease. There is a reported mortality of 20% to 25% for AHS. Studies in Han Chinese residing in Taiwan showed that the HLA-B*5801 allele was present in 100% of the patients with allopurinol-induced skin reactions, but in only 15% of allopurinol-tolerant controls. The 2012 American College of Rheumatology Guidelines for Management of Gout recommend that HLA-B*5801 testing should be considered in select patient subpopulations at an elevated risk for AHS. Those of Korean descent, especially those with stage 3 or higher chronic kidney disease, or of Han Chinese or Thai extraction irrespective of renal function, should be tested. Consideration should be given to testing all patients who will be given allopurinol.
The Food and Drug Administration recommends that individuals of Asian ancestry be genotyped for the presence of the HLA-B*1502 allele before receiving any of the following drugs: carbamazepine, a drug used to treat epilepsy, manic/bipolar disorders, and neuropathic pain; and phenytoin and its soluble precursor fosphenytoin, drugs used to control seizures. Some studies have also suggested a connection between HLA-B*1502 positivity and adverse reactions to lamotrigine used for treating epilepsy and bipolar disorder.
See www.mayocliniclabs.com for details of specimen collection.
Medications.
The use of antibacterial sulfonamides, anticonvulsant agents, oxicam nonsteroidal antiinflammatory drugs (NSAIDs), and allopurinol is associated with large increases in the risk of SJS or TEN (see Box 18.3 ). The excess risk of these drugs does not exceed 5 cases per million users per week. The risk of developing TEN from antiepileptic drugs is highest in the first 8 weeks after onset of treatment.
Prodromal Symptoms.
Fever is the most frequent prodromal symptom. Symptoms suggestive of an upper respiratory tract infection, such as headache and sore throat, usually precede the appearance of skin lesions by 1 or 2 weeks. Stomatitis, conjunctivitis, and pruritus occur 1 to 2 days before the onset of the rash.
Skin.
TEN begins with diffuse, hot erythema covering wide areas. In hours the skin becomes painful, and with slight thumb pressure the skin wrinkles and slides laterally and over the basal layer ( Fig. 18.9 ). This ominous sign (Nikolsky sign) heralds the onset of a life-threatening event. Small blisters and large bullae may appear. Nonerythematous skin usually remains intact, and the scalp is spared.
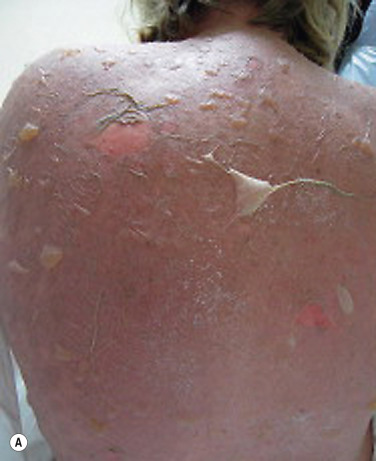
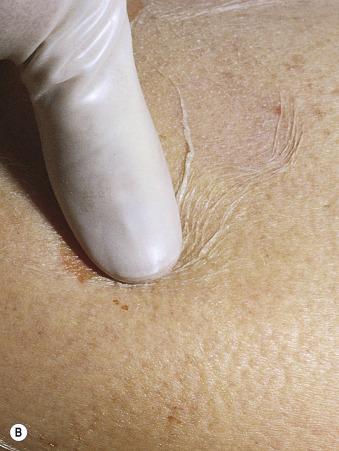
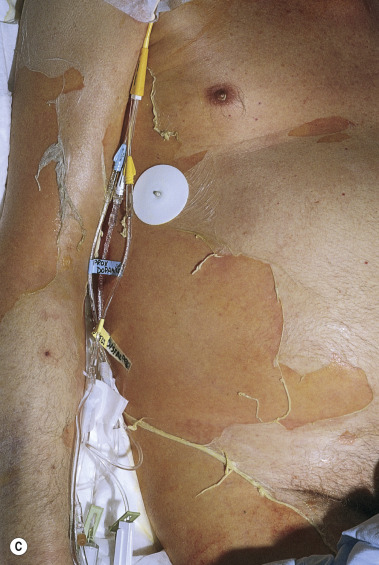
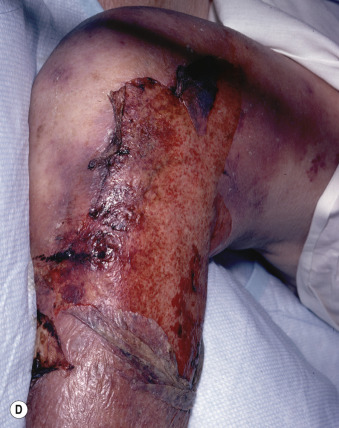
Blisters form and sheets of epidermis are sloughed, leaving weeping dermis. Epidermal detachment may progress for 5 to 7 days before reepithelialization occurs (typically 1 to 3 weeks). Keratinocyte apoptosis is seen on biopsy specimens.
Mucous Membranes.
Inflammation, blistering, and erosion of the mucosal surfaces, especially the oropharynx, are early and characteristic findings. The vaginal tract epithelium frequently blisters and erodes. Pain and erosion of oral mucous membranes interfere with oral intake, and nasogastric or duodenal tube feeding is often required. The remainder of the GI tract functions normally if sepsis does not occur.
Eyes.
Severe eye involvement is a constant feature. Purulent conjunctivitis leads to swelling, crusting, and ulceration with pain and photophobia. Complications include conjunctival erosions with subsequent revascularization, fibrous adhesions, and corneal ulceration and blindness. Photophobia, mucinous discharge, and decreased visual acuity may last for years.
Respiratory Tract.
Involvement of bronchial epithelium was noted in 27% of cases and must be suspected when dyspnea, bronchial hypersecretion, normal chest radiograph, and marked hypoxemia are present during the early stages of TEN. Bronchial injury indicates a poor prognosis. Life-threatening acute respiratory decompensation requiring ventilatory support and long-term pulmonary function abnormalities may occur. Patients should be closely monitored for pulmonary complications. Bronchopneumonia occurs in 30% of reported cases and is the cause of death in many cases. Many patients require intubation or ventilatory support. Respiratory failure can occur, with mucus retention and sloughing of the tracheobronchial mucosa.
Infection.
Septicemia and Gram-negative pneumonia are the most common causes of death. The lungs and denuded skin are the common portals of entry. The incidence of positive blood culture results is very high when central venous lines are used. Intravenous lines are changed or discontinued if it is probable that they are the source of positive blood culture findings. The urethra is often involved, but the use of Foley catheters can be avoided in many cases.
Fluid and Electrolyte Loss.
Fluid loss in TEN is not as severe as it is in burn patients, but significant losses can occur if grafts are not applied. Apparently, the acute-phase reactants that create massive edema after thermal injury are not released in TEN.
Other Complications.
Leukopenia of uncertain cause may occur. Toxins (e.g., from absorbed silver sulfadiazine) or immune complexes may be the cause. In one series, renal involvement consisting of hematuria, proteinuria, and elevated serum creatine level occurred in 50% of patients with SJS.
Prognosis.
SCORTEN (see Table 18.5 ) is an accurate predictor of mortality. Age older than 40 years, the presence of metabolic syndrome and/or gout, higher body surface area involvement, higher SCORTEN, and higher number of medical comorbidities statistically increased risk of death sigificantly.
Treatment
Stop the offending drug as soon as possible and consider referring the patient to a burn unit. Box 18.5 shows a general outline for a representative treatment protocol for a patient with SJS and TEN.
Protocol Elements
Identification and withdrawal of the culprit drug and all nonessential medications
Transfer of the patient to intensive care, burn unit, or other specialty unit
Supportive care
Thermoregulation
Airway protection
Fluid replacement and assessment of fluid balance
Nutritional support
Pain management
Venous thromboembolism prophylaxis
Monitoring for infection
Psychologic support
Medical treatment
Systemic immunomodulatory treatment: adults/children
Skin treatment
Mucous membrane treatment: ocular, oral, genital
Special considerations in pregnant women
Avoidance of high-risk drugs and monitor for secondary cutaneous adverse drug reactions
Monitoring and treatment of acute complications
Communication with the patient and family, health care providers, and regulatory agencies
Prior to discharge
Information on the diagnosis and the culprit drug
Medical recommendations: medical treatment and sun protection
Explanation of the possible long-term medical complications and medical follow-up
Offer of emotional support: patients should be asked to complete the GHQ-12 as a quick and valid screening instrument for psychologic distress; scores ≥2 should trigger a referral to a psychiatrist or psychologist
Referral to a support group
GHQ-12, General Health Questionnaire-12.
Systemic Steroids.
The use of systemic steroids is still controversial, but most authors recommend avoiding systemic corticosteroids.
Intravenous Immunoglobulin (IVIG).
Evidence favors the use of high-dose intravenous immunoglobulin (IVIG) (3 to 4 g/kg) for the treatment of TEN in some reports. High-dose IVIG does not significantly alter mortality.
Cyclosporine.
Patients with TEN treated with cyclosporin A (3 to 4 mg/kg per day) without other immunosuppressive agents experience rapid reepithelialization and improved survival. This regimen appears to be more effective than cyclophosphamide and corticosteroids.
Etanercept.
A single subcutaneous injection of 50 mg of etanercept may stop skin sloughing and promote reepithelialization (median healing time 8.5 days) without side effects.
Plasma Exchange.
Plasma exchange produced complete remission in two series. Plasmapheresis is a safe intervention in extremely ill patients and may reduce the death rate.
Burn Center Treatment.
TEN has pathophysiologic similarities to partial-thickness burn injury. The management of major fluid and electrolyte derangements, the intensive nutritional support, and the management of extensive cutaneous injuries with ready access to biologic and semisynthetic wound dressings are best accomplished in a multidisciplinary burn center.
The current trend toward prolonged treatment in outside facilities before referral to a burn center is detrimental to the care of patients with TEN. The overall rate of bacteremia, septicemia, and mortality is significantly reduced with early (≤7 days) referral to a regional burn center.
Separation at the junction of the dermis and epidermis leaves totally viable dermis and intact skin appendages. If the dermis can be protected from toxic detergents, salves, or desiccation, rapid resurfacing by proliferation of epithelium from the skin appendages will occur in approximately 14 days without scarring. Silver sulfadiazine and mafenide acetate (Sulfamylon) delay epithelialization.
Erythema Nodosum
Erythema nodosum (EN) is a nodular erythematous eruption that is usually limited to the extensor aspects of the extremities. EN represents a hypersensitivity reaction to a variety of antigenic stimuli and may be observed in association with several diseases (infections, immunopathies, malignancies) and during drug therapy (with halides, sulfonamides, oral contraceptives). Approximately 55% of cases are idiopathic. Laboratory tests show no specific abnormalities except those related to an underlying disease. Familial EN is reported, with affected family members showing a common haplotype. The incidence has decreased in the antibiotic era. EN is seen more frequently in females. The peak incidence occurs between the ages of 18 and 34 years. The female-to-male ratio is 5 : 1.
Clinical Manifestations and Course.
Prodromal symptoms of fatigue and malaise or symptoms of an upper respiratory tract infection precede the eruption by 1 to 3 weeks. The clinical picture is that of a nonspecific systemic illness, with low-grade fever (60%), malaise (67%), arthralgias (64%), and arthritis (31%). Pulmonary hilar adenopathy may develop as part of the hypersensitivity reaction of EN and is seen in cases with diverse causes.
Joint Symptoms.
Arthralgia occurs in more than 50% of patients and begins during the eruptive phase or precedes the eruption by 2 to 8 weeks. Symptoms may disappear in a few weeks or persist for 2 years, but they always resolve without destructive joint changes. Testing for rheumatoid factor is negative. Joint symptoms consist of erythema, swelling, and tenderness over the joint, sometimes with effusions; arthralgia and morning stiffness, most commonly in the knee, but any joint may be affected; and polyarthralgia lasting for days.
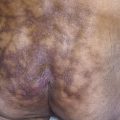
Skin Eruption.
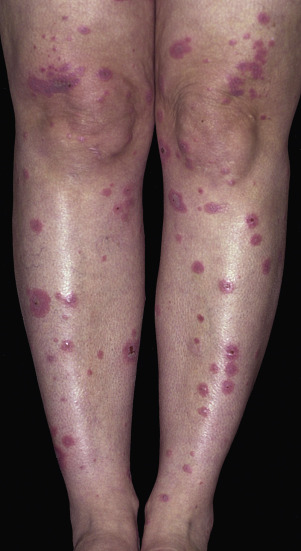
The eruptive phase begins with flu-like symptoms of fever and generalized aching. The characteristic lesions begin as red, node-like swellings over the shins; as a rule, both legs are affected. Similar lesions may appear on the extensor aspects of the forearms, thighs, and trunk ( Fig. 18.10 ). The border is poorly defined, with size varying from 2 to 6 cm. Lesions are oval, and their long axis corresponds to that of the limb. During the first week the lesions become tense, hard, and painful; during the second week they become fluctuant, as in an abscess, but never suppurate. The color changes in the second week from bright red to bluish or livid; as absorption progresses, it gradually fades to a yellowish hue, resembling a bruise; this disappears in 1 or 2 weeks as the overlying skin desquamates. The individual lesions last approximately 2 weeks, but new lesions sometimes continue to appear for 3 to 6 weeks. Aching of the legs and swelling of the ankles may persist for weeks. The condition may recur for months or years.