Lerner grading system [11]
Horn grading system [7]
Grade 0
Normal skin or unrelated cutaneous disease
Grade 1
Vacuolar alteration
Vacuolar alteration
Grade 2
Spongiosis and dyskeratosis (eosinophilic bodies)
Epidermal or follicular dyskeratotic cells, dermal lymphocytic infiltration
Grade 3
Epidermolysis and formation of bulla
Formation of subepidermal clefts and microvesicles
Grade 4
Total epidermal denudation
Epidermal separation from dermis
The histologic diagnosis of GVHD can be limited by findings suggestive of concurrent infection or drug eruption, by recent chemotherapy or radiotherapy, or by minimal foci to suggest unequivocal GVHD.
Acute GVHD is characterized histologically by basal cell vacuolar alteration, and may be accompanied by varying degrees of a lymphocytic lichenoid infiltrate [12]. In contrast, numbers of Langerhans cells (epidermal antigen-presenting cells) are reduced [13]. In early aGVHD, the vacuolar change may be very subtle, and the dermis often features only a sparse perivascular lymphocytic infiltrate (Fig. 5.1) [14]. During this stage, orthokeratosis, parakeratosis (apoptotic, cornified cells with retained nuclei), or both can be seen overlying a hypergranular layer. As the clinical severity of aGVHD progresses, necrotic or dyskeratotic keratinocytes (Fig. 5.2) are more frequently found and may be present in any epidermal layer, including the follicular epithelium (Fig. 5.3) [1, 15, 16]. Cytoid bodies, or “eosinophilic bodies” (i.e., necrotic keratinocyte debris), are often present either within the affected epidermis or in the immediately subjacent papillary dermis [7]. Lymphocytic exocytosis and satellite cell necrosis (lymphocytes in close apposition to apoptotic keratinocytes) are inconsistent features [16, 17]. There may frequently be a background of cytotoxic effects including loss of epidermal polarity with dysmaturation and atypical mitoses, often resembling low-grade squamous dysplasia; these cutaneous toxic effects of chemotherapy often partially limit interpretation, as cytotoxic agents may also produce necrotic keratinocytes [14]. Spongiosis is variable, and microvesiculation can occur [14, 18]. Vascular proliferation has been described in association with perivascular edema, a lymphocytic infiltrate, and prominent mast cells [19, 20]. On direct immunofluorescence, deposition of C3 and IgM at the dermal-epidermal junction is present in up to 39 % of aGVHD [21].
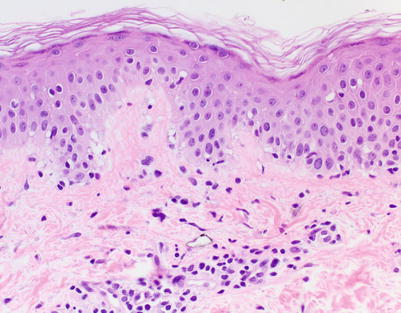
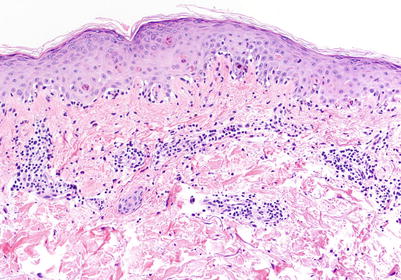
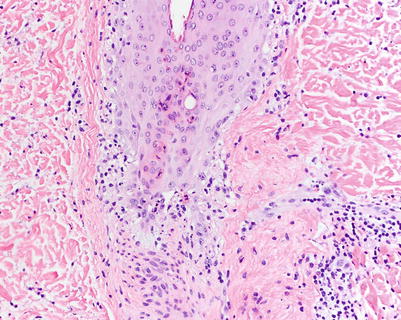
Fig. 5.1
Acute vacuolar interface dermatitis. Basal keratinocyte vacuolization is present, with cytotoxic lymphocytes aligned along the basal layer. Overtly necrotic keratinocytes are absent. These findings correspond to acute graft-versus-host disease (aGVHD) histologic grade I, changes considered insufficiently specific to make a definitive GVHD diagnosis (H&E-stained section, 200× magnification)
Fig. 5.2
Interface dermatitis with necrotic keratinocytes at and above the epidermal basal layer, corresponding to aGVHD histologic grade II (H&E, 100× magnification)
Fig. 5.3
Vacuolar interface change with necrotic keratinocytes within follicular epithelium in aGVHD (H&E, 200× magnification)
Clues to Histologic Diagnosis
Definitive diagnosis of aGVHD is more readily made when histologic Grade 3 or 4 changes are identified (Fig. 5.4). In less severe cases, given the lack of pathognomonic findings, histologic diagnosis of aGVHD can be difficult. However, there are several features that when taken into constellation may help in the diagnosis of aGVHD.
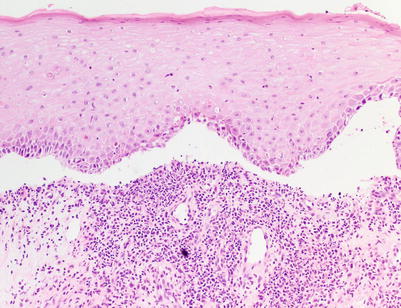
Fig. 5.4
Interface dermatitis (lichenoid type) with subepidermal clefting, corresponding to aGVHD histologic grade III (H&E, 100× magnification)
Though interface dermatitis may be encountered in GVHD, drug hypersensitivity, and viral exanthema (all clinical considerations in the acute post-transplant period), vacuolar alteration of adnexal structures is uncommon in cutaneous pathology and is not typically seen in most drug eruptions [22]. GVHD represents a prime example in which vacuolar alteration of adnexae, principally of follicular units, may be encountered (Fig. 5.3) [23]. However, adnexal vacuolar change also may be found in systemic lupus erythematosus (SLE) and toxic erythema of chemotherapy (TEC). Theoretically, it may also be found in dermatomyositis, but this has not been consistently reported in the literature. Because of the uneven distribution of cutaneous adnexal structures, level sections should be obtained to search for follicular units and sweat gland epithelium when there is a high clinical index of suspicion for aGVHD.
In addition to involvement of the follicular epithelium, diagnosis of aGVHD is more suggestive in the absence of eosinophils. However, the presence of eosinophils does not completely exclude a diagnosis of aGVHD until more than 16 eosinophils per 10 high power fields (HPF) are found (Fig. 5.5) [24]. The presence of >16 eosinophils per 10 HPF is 100 % specific for a cutaneous drug eruption, whereas >3.5 eosinophils per 10 HPF has 93 % specificity. In the context of aGVHD, a specificity of 93 % is likely inadequate, as a false negative diagnosis can result in significant morbidity and mortality. However, finding at least 4 eosinophils per 10 HPF should prompt consideration of “possible GVHD” rather than “consistent with or highly suggestive of GVHD” as the final diagnosis.
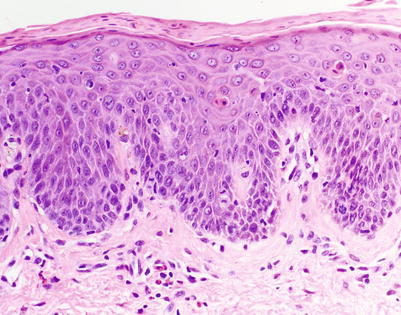
Fig. 5.5
Interface dermatitis with perivascular eosinophils. These findings may be seen in aGVHD, but the presence of more than 4 eosinophils per 10 HPF around the superficial perivascular plexus makes a drug hypersensitivity reaction more likely. A diagnosis of “possible GVHD” with a comment regarding tissue eosinophilia is likely the most appropriate approach in this setting (H&E, 200× magnification)
Differential Diagnosis of Acute GVHD
The histologic features of aGVHD are mimicked by several entities (Table 5.2). The microscopic features of aGVHD are largely identical to engraftment syndrome (and likely pre-engraftment syndrome), which requires clinicopathologic correlation. Unlike aGVHD, however, engraftment syndrome typically occurs 10–14 days after transplantation, specifically 48 hours before or after the first appearance of neutrophils in peripheral blood (absolute neutrophil count >500) [25]. This event usually precedes the appearance of peripheral lymphocytes.
Table 5.2.
Histologic differential diagnosis of acute graft-versus-host disease
aGVHD | ES | TEC | SJS/TEN | |||
|---|---|---|---|---|---|---|
Epidermis | Keratinocytes | Necrotic | +a | + | + | + |
Atypical | + | |||||
Lymphocytic exocytosis | + | + | ||||
Basal vacuolization | + | + | + | + | ||
Dermis | Perivascular lymphocytes | + | + | ± | ||
Eosinophils | ± | + | ||||
Neutrophils | ± | |||||
Edema | + | + | ||||
Adnexae | Vacuolar alteration | + | + | + | ||
Necrotic keratinocytes | + | + | ||||
Peri-eccrine neutrophils | + | |||||
Squamous syringometaplasia | + | |||||
Similar to aGVHD, toxic erythema of chemotherapy (TEC) can also present in the post-transplantation period with necrotic keratinocytes involving the epidermis and adnexal structures (Fig. 5.6), especially eccrine epithelium (likely owing to the fact that chemotherapy may concentrate in eccrine sweat). Significant keratinocyte dysmaturation and/or cytologic atypia leans toward the diagnosis of TEC [22]. In addition, TEC also may show dermal edema, eccrine squamous syringometaplasia, and peri-eccrine neutrophils [26]. Theoretically, TEC is a direct toxicity effect, a nonimmunologic process that should be minimally inflammatory, but in practice, biopsies often demonstrate mild perivascular lymphocytic inflammation that does not differ significantly from that seen in many aGVHD cases.
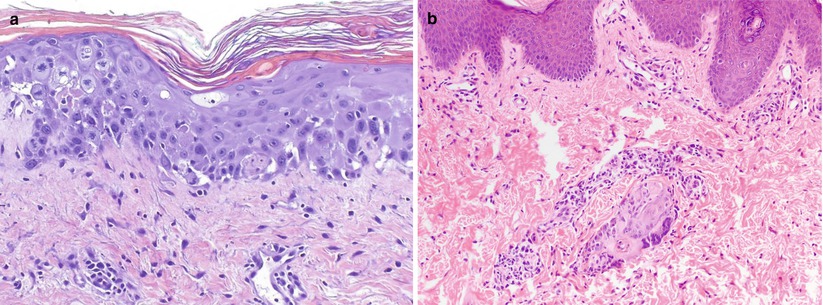
Fig. 5.6
Toxic erythema of chemotherapy. (a) Atypia of epidermal keratinocytes and multiple mitotic figures, resulting from accumulation of cytotoxic agents within the skin. Note basal layer vacuolization and necrotic keratinocytes mimicking aGVHD (H&E, 200× magnification). (b) Interface dermatitis with focal necrosis isolated to eccrine sweat duct epithelium only. The absence of overlying epidermal changes is a clue to the diagnosis in this specimen (H&E, 100× magnification)
As discussed under clues to diagnosis, drug hypersensitivity reactions such as morbilliform drug eruption are favored in the presence of eosinophils, with >16 eosinophils per 10 HPF ruling out aGVHD [24]. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis spectrum disorders may be indistinguishable from aGVHD, but involvement of adnexal structures and involvement of follicular epithelium is more likely in aGVHD, as noted above. In addition, squamatization of the basal layer and parakeratosis or compact hyperkeratosis are more likely to be seen in aGVHD than in erythema multiforme [27]. The presence of bile pigment has been suggested as a marker to differentiate aGVHD from erythema multiforme, though its sensitivity is only 6 % [28].
Viral exanthems may also commonly mimic aGVHD microscopically, with basal layer vacuolization seen in up to 40 % of cases [29]. Some histopathologic clues may provide a specific viral diagnosis in limited cases. Multinucleate keratinocytes are suggestive of herpes simplex or varicella infection. Human herpes virus-6 may show distinctive viral inclusions within lymphocytes: a halo surrounds the irregularly shaped nuclei, which contain a central basophilic inclusion [30]. Cytomegalic endothelial cell nuclei, or “owl’s eyes,” may be found in dermal blood vessels in cytomegalovirus infections [31]. Serum polymerase chain reaction (PCR) studies can be performed for confirmation when skin biopsies suggest viral infection.
Other interface dermatoses must also be excluded from the differential. Acute systemic lupus erythematosus (SLE) may be the most difficult to distinguish from aGVHD, with each revealing a sparse lymphocytic interface dermatitis, though SLE is not a common clinical consideration in the period immediately after transplantation. Findings of increased mucin in the superficial dermis may help to distinguish the two [32]. In discoid lupus or other lesions of chronic cutaneous lupus, more prominent superficial and deep lymphocytic infiltrates, follicular plugging, and increased dermal mucin deposition can be seen. Dermatomyositis may be distinguishable from GVHD only by the presence of increased dermal mucin and/or C5b-9 deposition at the dermo-epidermal junction on direct immunofluorescence [33]. Both pityriasis lichenoides et varioliformis acuta (PLEVA) and pityriasis lichenoides chronica (PLC) reveal a sparse vacuolar interface in early lesions. As PLEVA continues to develop, vacuolar change becomes more extensive, and parakeratosis, a dense wedge-shaped lymphocytic infiltrate, and prominent erythrocyte extravasation may be found [34]. Within the parakeratosis, there may be an inflammatory infiltrate composed of neutrophils or sometimes lymphocytes. Unlike PLEVA, PLC is more likely to have broad parakeratosis without neutrophils and with fewer necrotic keratinocytes.
Potential Adjunctive Tests
Given the significant adverse outcomes that can occur in untreated aGVHD, several tools have surfaced as aids in the diagnosis of GVHD. HLA immunostaining is utilized in some centers, with HLA-DR staining of keratinocytes found to be up to 100 % specific [13]. Significant recent research has focused on identifying biomarkers of prognostic and diagnostic significance in GVHD. Biomarker assays commonly utilize serum, but they can also be performed on tissue via immunostains or cytogenetic tests.
The biomarker thymic stromal lymphoprotein (TSLP) may be helpful in predicting which patients are likely to develop aGVHD. TLSP is a keratinocyte-derived cytokine that skews the immune response toward a Th2 phenotype. In cutaneous specimens taken 20 to 30 days after transplantation, elevated TSLP has been found in patients who later develop aGVHD [35]. TLSP levels were not elevated in patients who did not develop cutaneous aGVHD. A TSLP immunostain is available as an investigational assay and labels epidermal keratinocytes.
Of the biomarkers studied, elafin, an elastase inhibitor expressed by the inflamed epidermis, has emerged as a reliable marker of aGVHD activity [36, 37]. Evaluation of elafin can be performed on both serum and histologic skin specimens. It is unclear whether elafin levels can help differentiate aGVHD from other diagnoses, but once a diagnosis of aGVHD has been unequivocally made, measurement of elafin levels via serum ELISA can help prognosticate disease; increased elafin levels portend a poorer prognosis [37].
Given the utility of elafin, it has recently been included in a six-marker panel in the evaluation of aGVHD [38]. Also included in this panel were previously validated diagnostic biomarkers IL-2 receptor-α, tumor necrosis factor receptor-1, hepatocyte growth factor, IL-8, and regenerating islet–derived 3-α (reg3α). The six-biomarker panel was able to predict two clinical outcomes: which patients would be nonresponsive at day 28 post-therapy, and mortality risk at day 180 from aGVHD onset. The use of this biomarker panel may aid in determining patients at high risk for treatment nonresponse and mortality, supporting the potential for earlier intervention and change in therapeutics.
Special Situations
In patients with hematologic malignancies, the use of T-cell–depleted (TCD) bone marrow transplants is an effective method of reducing the risk of GVHD [39]. Similarly to T-cell–replete transplant recipients (TCR) who develop aGVHD, TCD patients who develop aGVHD share conserved histologic features [40]. In these patients, a diagnosis of aGVHD was made on the findings of diffuse basal layer vacuolar alteration, more than three necrotic keratinocytes per HPF, and necrosis involving all layers of the epidermis. In contrast to TCR patients, however, TCD patients with aGVHD were more likely to have follicular involvement (77 % vs 16 %), dermal eosinophils (31 % vs 3 %), dermal neutrophils (31 % vs 0 %), satellitosis (77 % vs 24 %), lymophocyte exocytosis (92 % vs 37 %), and extravasated erythrocytes (69 % vs 13 %). The significance of these variations has not been determined, and further studies are needed to assess whether clinical outcomes are correlated with these histologic differences.
Recently, children with dystrophic and junctional epidermolysis bullosa have been treated with stem cell transplants. From October 2007 to August 2009, seven children with recessive dystrophic epidermolysis bullosa were treated with a co-infusion of mesenchymal stromal cells and hematopoietic stem cells [41]. None of the seven patients developed acute or chronic GVHD in up to 2 years of follow-up. It is unclear why these patients have a decreased GVHD incidence. It has been suggested that the decreased incidence of GVHD may be from the presence of mesenchymal stromal cells or from an inherent defect in the skin of patients with recessive dystrophic epidermolysis bullosa [42]. Further histopathologic evaluation and studies may be helpful in the development of tools or conditioning regimens to prevent GVHD onset.
Chronic Graft-Versus-Host-Disease
Broad diagnostic criteria make it difficult to estimate the exact incidence and prevalence of cGVHD. The NIH Pathology Working Group is striving to create more uniform criteria to help establish the diagnosis. At this time, it is estimated that the prevalence of cGVHD is approximately 50 % [1, 43]. The skin is the most frequent organ site to be involved in cGVHD, with cutaneous disease occurring in up to 75 % of patients at the time of diagnosis [44]. The presence of at least one diagnostic clinical feature of cutaneous cGVHD negates the need for biopsy (Table 5.3) [1]. Biopsy is recommended to help confirm the diagnosis of cGVHD in the presence of only distinctive clinical features—those that are seen in cGVHD but are insufficient as isolated findings to definitively diagnose cGVHD. In the skin, the most common distinctive feature is depigmentation, but other features that may be present include hypopigmentation, hyperpigmentation, ichthyosis, keratosis pilaris, or sweat impairment (hypohidrosis) [1]. Other distinctive features involving the nails, hair, oral mucosa, and genitalia are discussed in further detail in the clinical chapters of this book.
Cutaneous | Oral mucosa | Genitalia | |
|---|---|---|---|
Poikiloderma | + | ||
Lichen planus–like features | + | + | + |
Sclerotic features | + | +a | + |
Morphea-like features | + | ||
Lichen sclerosus–like features | + | ||
Hyperkeratotic plaques | + |
Biopsy can be an invaluable tool in confirming suspicion for cGVHD. The size of the biopsy specimen is dependent on the clinical picture. For patients with non-sclerotic cGVHD, the specimen should be at least 4 mm in size, preferably from a palpable lesion without overlying secondary change [3]. In sclerotic lesions, an excisional biopsy is preferred. However, a punch biopsy of 6–8 mm is also an acceptable alternative [3]. Biopsy of a bullous or vesicular lesion should be done at the edge of the blister and should include surrounding erythema. Although biopsy can be a significant diagnostic aide, its use in long-term follow-up to assess treatment response has not been validated.
Main Histologic Patterns in cGVHD: Lichenoid and Sclerodermoid
Classically, chronic cutaneous GVHD has been clinically and histologically classified into two subtypes: an early lichenoid stage and a later sclerotic stage. In early cGVHD, patients clinically present with lichen planus-like papules with histopathologic findings nearly identical to aGVHD (Fig. 5.7) [45]. The epidermis shows irregular acanthosis, parakeratosis, hypergranulosis, vacuolar alteration, and variable keratinocyte necrosis (again, a key feature of GVHD). A bandlike lymphocytic infiltrate may obscure the dermal-epidermal junction with evidence of basal vacuolar degeneration, though many biopsies of lichenoid GVHD demonstrate a dermal lymphocytic infiltrate that is significantly sparser than lichen planus and other lichenoid dermatitis. Squamous eccrine metaplasia can be seen in the transition from acute to chronic GVHD [46].
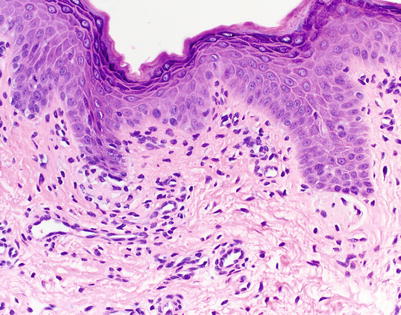
 Grading and Treatment of Acute Graft-Versus-Host Disease
Grading and Treatment of Acute Graft-Versus-Host Disease
 Wound Care in the Management of Chronic Graft-Versus-Host Disease
Wound Care in the Management of Chronic Graft-Versus-Host Disease
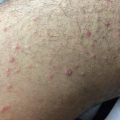 Clinical Presentation of Acute Cutaneous Graft-Versus-Host Disease
Clinical Presentation of Acute Cutaneous Graft-Versus-Host Disease
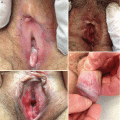 Clinical Presentation of Mucosal Acute and Chronic Graft-Versus-Host Disease
Clinical Presentation of Mucosal Acute and Chronic Graft-Versus-Host Disease
 Dermal and Subcutaneous Chronic Graft-Versus-Host Disease
Dermal and Subcutaneous Chronic Graft-Versus-Host Disease
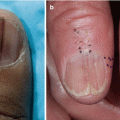 Clinical Presentation of Nonsclerotic Epidermal Chronic Graft-Versus-Host Disease and Hair and Nail Changes
Clinical Presentation of Nonsclerotic Epidermal Chronic Graft-Versus-Host Disease and Hair and Nail Changes




Related posts:
Grading and Treatment of Acute Graft-Versus-Host Disease
Wound Care in the Management of Chronic Graft-Versus-Host Disease
Clinical Presentation of Acute Cutaneous Graft-Versus-Host Disease
Clinical Presentation of Mucosal Acute and Chronic Graft-Versus-Host Disease
Dermal and Subcutaneous Chronic Graft-Versus-Host Disease
Clinical Presentation of Nonsclerotic Epidermal Chronic Graft-Versus-Host Disease and Hair and Nail Changes
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
