Diagnostic
Skin Biopsy, preferably excisional sent for: Histology Immunohistochemistry
Evaluation
Baseline studies of complete blood count (CBC), complete metabolic panel (CMP), gamma glutamyl transferase (GGT), INR/PT, APTT/PTT, fibrinogen, early morning urine specific gravity and osmolality, abdominal ultrasound, chest radiograph, and skeletal survey. PET scan useful for scanning for extracutaneous lesions if available
Specialized testing including MRI, high resolution CT, bone marrow biopsy, and lung biopsy as indicated based on results of baseline evaluation and clinical scenario. Multispecialty involvement recommended in such circumstances
Treatment
Methotrexate: Pregnancy test (for women of child-bearing potential), CBC, CMP, tuberculosis testing at baseline, monitor CBC and hepatic panel during treatment
Retinoids: Pregnancy test (for women of child-bearing potential), CBC, AST, ALT, fasting lipids at baseline and during treatment
Thalidomide: Pregnancy test (for women of child-bearing potential), CBC. Monitor CBC and pregnancy tests during treatment
Azathioprine: Thiopurine methyltransferase (TPMT), pregnancy test (for women of child-bearing potential), CBC, CMP, tuberculosis testing at baseline. Monitor CBC and CMP during treatment
Diagnosis requires histologic and immunophenotypic identification of the characteristic CD1a+, CD207+ langerhans cells in the appropriate clinical setting, and is greatly aided by biopsy of an involved skin lesion. Referral for curettage of the center of an involved bone lesion may also yield the diagnosis.
Prior to initiating treatment, a complete history and physical examination should be performed, with attention paid to a review systems of pain, swelling, rashes, otorrhea, irritability, fever, loss of appetite, diarrhea, weight loss or poor weight gain, growth failure, polydipsia, polyuria, change in energy level, and behavioral or neurological changes. Baseline staging studies recommended by expert consensus panel include laboratory and radiologic studies to evaluate for systemic involvement [12]. PET scan can be helpful for baseline identification of extracutaneous LCH lesions, though availability may be limited due to cost. More specialized testing can be pursued based on the results of the screening studies and clinical scenario.
Close observation | D |
Topical steroids, moderate and high potency | Ea |
Given the often self-resolving or limited course of skin-limited disease, close observation alone is recommended unless skin lesions are symptomatic. In children with a more rash-like eruption, moderate- to high-potency topical steroids may be used, though benefit is typically marginal [12].
Nitrogen mustard has been shown to be effective in treatment of cutaneous LCH lesions in children [13]. Treatment may be limited by contact dermatitis to the agent, and treatment must be weighed against potential mutagenic effect, though fortunately there have been no reports of secondary tumors developing secondary to nitrogen mustard use in LCH [12]. One recent nitrogen mustard regimen reported to be effective was 20 mg mechlorethamine hydrochloride dissolved in 40 ml water applied once daily for 14 days, followed by taper to daily use for 1 week per month and subsequently twice per month as lesions regressed [13]. Other treatments that have been reported to offer benefit include nbUVB and topical imiquimod cream [10, 14]. In the adult literature, imiquimod applied five times/week for 2 months was beneficial in one reported case [15]. For symptomatic nodules, surgical excision may be appropriate in some circumstances, though radical excision should be avoided.
For symptomatic skin involvement causing significant morbidity that is refractory to local treatment modalities, oral corticosteroids and low-dose methotrexate can be considered. A reported pediatric methotrexate regimen is 20 mg/m2 weekly, and the successful combination of this with alternating day dosing of prednisone 40 mg/m2/day has been reported [11, 16]. Thalidomide at a dose of 50 mg daily for children has been shown to be effective in low-risk skin disease, though it must be weighed against a significant toxicity profile including fatigue, neutropenia, peripheral neuropathy, and increased risk of deep-vein thrombosis and thromboembolism [17]. Successful use of photodynamic therapy with methylaminolevulinate has been reported in an infant [18]. Oral retinoids, such as acitretin, azathioprine, interferon alpha 2, and PUVA therapy have been anecdotally reported to be effective in adults, and can be considered in children in severe cases [10, 12, 19]. Radiation therapy is generally not recommended given concern for long-term sequelae in children, though it is very effective for treatment of painful ulcerated lesions refractory to other treatments [20].
Juvenile Xanthogranuloma
Clinical Features
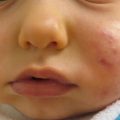
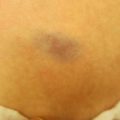
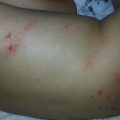
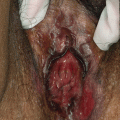
Juvenile xanthogranuloma (JXG) is the most common non-Langerhans cell histiocytosis. It is benign, self-limited, and presents in infants and children more commonly than adults. JXG is derived from dermal dendrocytes and presents in normolipemic individuals without abnormalities in lipid metabolism. Cutaneous lesions often present on the head, neck, and trunk and can be divided by their size into ‘micronodular’ (lesions <1 cm) and ‘macronodular’ (lesions >1 cm) forms. Giant JXG lesions are also rarely seen, and can be up to 5–10 cm in size. The lesions are often asymptomatic, but can ulcerate and bleed. Clinically, they present as solitary to numerous firm, round papules or nodules. The solitary presentation is more common, occurring in up to 90 % of all patients with JXG. Early on they present as more erythematous papules with minimal yellow-orange color. As they mature they become more characteristically yellow in color and may develop overlying telangiectasias. This correlates histologically to earlier lesions showing monomorphic nonlipid containing histiocytes with more mature lesions containing foamy cells and Touton giant cells with positive fat immunohistochemical staining. Extracutaneous involvement can occur, with ocular involvement being the most common, presenting in approximately 0.4 % of patients [21]. Other sites that have been reported include lungs, bone, kidneys, pericardium, colon, testes, ovaries, and liver [22]. Mucous membranes can be involved, but very rarely. JXG may be present at birth (30 %), but most cases present during the first year of life (75 %), with rare onset in adulthood reported. Onset is often abrupt with lesions continuing to persist or erupt for years, followed by spontaneous regression in 3–6 years time [23].
There has been as association seen between patients with JXGs and neurofibromatosis type 1 (NF1) [24] and JXGs and childhood leukemia, most commonly juvenile chronic myelogenous leukemia (JCML) [25]. In the majority of cases the JXG precedes or occurs at the same time as the leukemia is diagnosed and often multiple JXGs are present. There is also a triple association between JXG, NF1, and JCML in which individuals with JXG and NF1 are at a 20- to 32-fold higher risk of developing JCML than an individual with NF1 and no JXGs [26].
Investigation Recommendations
Diagnostic |
Skin biopsy for: |
Histopathology |
Immunohistochemistry |
Electron microscopy |
Evaluation |
Complete full body skin examination |
Complete review of systems |
Ophthalmology examination (if multiple JXGs) |
Diagnosis can be made clinically and can be confirmed by skin biopsy if needed. Skin biopsy and specific immunohistochemical stains show the following:
Histology: Nodular histiocytic infiltrate in the dermis sometimes with extension to upper subcutis. Early lesions are monomorphic and have few lipid-laden cells, where as more mature have foamy histiocytes and Touton giant cells (‘wreath’ of nuclei surrounded by foamy cytoplasm) and interstitial fibrosis. They can also be scattered neutrophils, rare plasma cells, and few eosinophils.
Immunohistochemical stains: Negative CD1a and S100 and positive Factor XIIIa and CD68
Electron microscopy: Reveals histiocytes with lipid-laden vacuoles, lysosomes, cholesterol clefts and myeloid bodies. Absence of Birbeck granules
A complete skin examination must be performed to determine the number of JXGs and rule out co-existent NF1. Patients that are 2 years of age or younger with multiple JXGs (2 or more) require an ophthalmology examination to rule out ocular involvement [21]. A complete review of systems should be performed to determine whether further evaluation should be undertaken to look for the rare circumstance of extracutaneous involvement.
Management Strategies
Juvenile xanthogranuloma is benign condition with spontaneous resolution of cutaneous and visceral lesions within 3–6 years. Cutaneous lesions can leave behind residual anetoderma, mild atrophy, and/or hyperpigmentation. Treatment is generally not indicated for cutaneous lesions given spontaneous resolution. However, treatment can be pursued in symptomatic cases or where there is significant cosmetic disfigurement.
On the other hand, in cases where there is intraocular involvement, there is high risk of morbidity that can result in spontaneous hyphema, glaucoma or blindness if left untreated [21]. Therefore, early treatment of ocular lesions is warranted.
Cutaneous Disease
Ocular Disease
Treatments that have been tried for ocular involvement, which can occur in the presence or absence of skin disease, include surgical resection for localized disease [29, 30], topical or systemic corticosteroids [29, 31], carbonic anhydrase inhibitors [29, 32], pilocarpine [29], and when medical management has failed radiation therapy [27, 29, 32].
Visceral Lesions
Visceral lesions do not usually need to be treated, as they do spontaneous resolve, unless there is interference with organ functionality or if they are causing symptoms. Treatments that have been used include chemotherapy, [27] corticosteroids, [27, 34] cyclosporine, [27] and radiotherapy [27, 34, 35].
Benign Cephalic Histiocytosis
Clinical Features
Benign cephalic histiocytosis (BCH) is a cutaneous, self-healing, non-Langerhans cell histiocytosis. Clinically, it is characterized by 2–6 mm small, asymptomatic, yellow-brown or red-brown macules or slightly elevated papules classically located on the face, less commonly located on the neck and trunk, and rarely on the extremities, buttocks, or genital region. Mucous membranes and acral surfaces are spared. BCH typically occurs prior to 6 months of age and generally before 3 years of age. Average age of onset is approximately 15 months. Onset of regression on average has been reported at about 23 months (range 8–48 months), with complete regression on average by 50 months (9–108 months) [36]. BCH may be considered to lie along a spectrum with other non-Langerhans cell histiocytosis such as juvenile xanthogranuloma (JXG) and generalized eruptive histiocytosis, with overlapping clinical and histologic features. There have been cases of BCH that have showed transformation to JXG after initial biopsy and diagnosis [37]. Some even consider BCH a variant of micronodular juvenile xanthogranuloma.
Investigation Recommendations
Diagnostic |
Skin biopsy for: |
Histopathology |
Immunohistochemistry |
Electron microscopy |
Diagnosis can be made clinically and can be confirmed by skin biopsy if needed. There can be overlap between the various histiocytic conditions. Some features that may help differentiate BCH from other histiocytoses that may have internal involvement and a more insidious course are as follows:
Histology: Well circumscribed histiocytic infiltrate in the superficial to mid-reticular dermis
Stains: Negative CD1a and S100 (can sometimes be weakly positive) and positive Factor XIIIa and CD68
Electron microscopy: Reveals intracytoplasmic comma-shaped or worm like bodies, coated vesicles, and desmosome like structures. Absence of Birbeck granules
Management Strategies
No treatment is necessary, as BCH runs a benign course with spontaneous healing. There can be residual macular dyspigmentation or atrophic scars. Since there can be overlap with other histiocytic entities, clinical follow-up for progression, review of systems, and physical examination for any internal involvement is recommended. Systemic disease has not been reported with BCH, but there has been one case of diabetes insipidus [38] and one case of insulin-dependent diabetes mellitus in a child with BCH [39].
Xanthoma Disseminatum
Clinical Features
Xanthoma disseminatum (XD) is a rare mucocutaneous non-Langerhans cell histiocytosis that occurs in both children and adults. There is predominance in males, and most cases have their onset prior to 25 years of age. XD can be classified into three forms by its evolution and prognosis: a self-healing form with spontaneous resolution; a more common persistent form in which lesions may never resolve; and a very rare progressive form with systemic involvement, organ dysfunction, and potential central nervous system (CNS) involvement [40].
Most patients have no alteration in lipid metabolism and have normal lipid profiles. Clinically, patients present with numerous small ovoid yellow-red to brown papules, plaques and nodules that occur predominantly on the face, flexural and intertriginous areas, around the umbilicus, perineum, and genital region. On the face the most prominent location is the eyelids. Approximately 30–50 % of cases have mucosal involvement including the oropharynx and gastrointestinal tract, conjunctiva and cornea, and upper respiratory tract (epiglottis, larynx, and trachea) [41]. Depending of the location, involvement of mucosal surfaces can result in dysphagia, dysphonia, visual impairment and blindness, respiratory distress, and intestinal obstruction. XD may also rarely manifest itself in the central nervous system, bone, liver, and within other ocular structures. Diabetes insipidus has been reported in about 40 % of patients with XD and tends to be transient, mild, and desmopressin responsive.
Investigation Recommendations
Diagnostic |
Skin biopsy for: |
Histopathology |
Immunohistochemistry |
Electron microscopy |
Evaluation for systemic involvement: |
MRI |
F-fluorodeoxyglucose PET/CT scan |
Diagnosis can be made by skin biopsy in conjunction with history and clinical appearance. Systemic involvement can rarely occur, therefore thorough physical examination, review of systems, and possible further imaging studies may be warranted to determine disease extent.
There can be overlap between the various histiocytic conditions. Some features that may help differentiate xanthoma disseminatum from other histiocytoses are as follows:
Histology: Histiocytic infiltrate in the dermis with presence of Touton and foreign body giant cells admixed with a mild inflammatory cell infiltrate of lymphocytes, plasma cells, and neutrophils.
Immunohistochemical Stains: Negative CD1a and S100 and positive for Factor XIIIa and CD68.
Electron microscopy: Lipid-laden histiocytes with prominent endoplasmic reticulum and fat droplets. Absence of Birbeck granules.
Given higher risk of systemic involvement, imaging is also recommended, including MRI. Recent data also supports the use of F-Fluorodeoxyglucose positron emission tomography/computed tomography (F-FDG PET/CT), which detects increased glucose metabolism, correlating to cell proliferation; this may detect systemic involvement earlier than MRI [42].
Management Strategies
Patients with XD should be followed for progression of disease or symptoms. There are no curative treatments for XD. Various treatment modalities have been reported in the literature as monotherapy or in combination; however, given the minimal data with these treatment modalities, there are none that are recommended as first-line, second-line, or third-line. As such, we will list them below as potential treatment options.
Surgery | E |
Ablative carbon dioxide laser | E |
Non-ablative diode laser | Ea |
Radiotherapy | E |
Corticosteroids | Ea |
Cyclophosphamide | E |
Azathioprine | E |
Chemotherapy (various regimens) | |
Lipid-lowering agents: combination of rosiglitazone, simvastatin, and acipimox or fenofibrate | Ea |
2-chlorodeoxyadenosine (cladribine) | Da |
Anakinra | Ea |
Various treatment modalities have been reported in the literature as monotherapy or in combination ranging from use of localized therapies such as surgery, ablative carbon dioxide lasers, [43] non-ablative, 1,450 nm laser, [44] and radiotherapy [45] directed at cutaneous and accessible lesions, lipid lowering medications, immunosuppression, and chemotherapy. The decision to pursue treatment is dependent of the extent of disfigurement, symptoms, anatomical location, and potential morbidity and mortality. Response rates to various treatment modalities are quite variable with no one modality showing consistent and successful results. Localized treatments are shown to be effective at treating individual lesions often without reoccurrence in the treated area, but they do not keep new lesions from developing and are limited by use for extensive disease. Corticosteroids have shown partial response in some cases, [46] but response has not been durable, with recurrence of lesions and progression of disease once corticosteroids were discontinued. Cyclophosphamide and azathioprine have been noted to have improvement in disease state in few cases [47]. There have been various chemotherapy regimens with variable response. Lipid-lowering agents have shown some effect, even in individuals with normal lipid profiles [48]. In particular, a regimen of rosiglitazone, simvastatin, and acipimox or fenofibrate have been used, which has shown some partial remission and stabilization of disease [49]. Use of 2-Chlorodeoxyadenosine (Cladaribine) has also shown some promising results in a case series of eight patients [50, 51]. 2-Chlorodeoxyadenosine is a purine analogue which inhibits adenosine deaminase enzyme and has been successfully used to treat numerous hematological malignancies and used in other systemic histiocytoses. Doses of 0.14 mg/kg/day for 5 days per month are repeated monthly for five to eight cycles. After three to five cycles, cessation of new lesions with significant improvement was noted in old lesions in all patients. Treatment was well tolerated without development of new lesions over follow-up period of 3 months to 8 years. There has also been a recent case report suggesting the use of a interleukin-1 receptor antagonist, Anakinra, with complete resolution of disease in a patient with cutaneous and CNS involvement [52]. This medication has been successfully used in other histiocytic disorders, such as multicentric reticulohistiocytosis and Erdheim-Chester disease.
Spontaneous resolution of XD lesions has also been reported, but most patients have the lesions lifelong. There have not been any reports of malignant transformation of cutaneous lesions.
Necrobiotic Xanthogranuloma
Clinical Features
Necrobiotic xanthogranuloma (NXG) is a rare disorder that is predominantly reported in adults. Clinically, it is characterized by asymptomatic or pruritic, well demarcated, yellow-orange to red-brown papules and nodules that coalesce to form an indurated plaque, often with overlying telangiectasias. Lesions have a predilection for periorbital areas, trunk, and the extremities. Ulceration, atrophy, and scarring can occur. Periorbital lesions and ocular involvement are very common and occur in 80–85 % of patients [53]. Systemic involvement can also occur in the heart, bone marrow, liver, spleen, larynx, pharynx, kidney, skeletal muscle, ovary, and intestine [54]. Oral mucosal erosions may also be present. There is often an associated underlying lymphoproliferative disorder, myelodysplastic disorder, or a paraproteinemia [53]. Paraproteinemia is found in 80–90 % of cases, with IgG kappa monoclonal gammopathy being the most common [55]. Lymphoproliferative and myelodysplastic disorders include multiple myeloma, chronic lymphocytic leukemia, and Hodgkin’s and non-Hodgkin’s lymphoma. NXG can occur an average of 2.5 years prior to the onset of the myeloma or myelodysplastic disorder.
Investigation Recommendations
Diagnostic |
Biopsy of skin or other affected organ |
Evaluation |
Laboratory evaluation: CBC with differential, comprehensive metabolic panel, lipid panel, serum protein electrophoresis, ESR and CRP |
Bone marrow biopsy |
Echocardiogram |
CT or MRI |
Ophthalmology examination |
Diagnosis can be confirmed by skin biopsy or biopsy of another affected organ in addition to clinical presentation.
Histology: Granulomatous inflammation located in the dermis with extension to the subcutaneous fat with large zones of necrobiotic collagen with characteristic cholesterol clefts with multinucleated and Touton giant cells and foreign body type cells with surrounding lymphocytic and plasma cell infiltrate.
Laboratory evaluation looking for lymphoproliferative disorders, other organ involvement, bone marrow dysfunction, and paraproteinemia should be performed. This may include CBC with differential, comprehensive metabolic panel, lipid panel, serum electrophoresis, ESR and CRP (often significantly elevated) [56].
Bone marrow biopsy may be indicated if there is a paraproteinemia or lab abnormalities. An echocardiogram should be done at baseline to determine whether there is cardiac involvement. Finally, imaging, including CT or MRI, may be indicated to evaluate the soft tissues, other organ involvement, or the orbit and ocular structures
Management Strategies
Patients with NXG should have regular continued follow-up to evaluate for systemic involvement and development of an underlying myelodysplastic syndrome, as these can occur an average of 2.5 years after the onset of the NXG. Follow-up should also include routine ophthalmologic evaluation. The course of NXG is chronic and indolent. The prognosis is generally good, with reports of 100 % patient survival at 10 years and 90 % survival at 15 years [57]. Prognosis of NXG largely depends on the extent of extracutaneous involvement.
There have been numerous reports of various treatments that have been tried, with variable outcomes. There are no consensus guidelines in regards to treatment due to the rarity of the disorder and lack of clinical and randomized control trials.
For limited cutaneous disease potent topical corticosteroids have shown little effect, but intralesional corticosteroids have proved to be more effective [58]. Ablative laser treatment with carbon dioxide (CO2) laser has been trialed, in addition to surgical excision for small localized lesions [58, 59]. With laser and surgical excision there have been reported recurrence of lesions. Other therapies that have been tried for cutaneous lesions are topical nitrogen mustard (mechlorethamine) [60], phototherapy (Psoralen ultraviolet A [PUVA]), [61] and localized radiation therapy [58] with some reported success.
Often this disorder does not present only with cutaneous disease, therefore systemic therapies are needed to help control and treat the disease. Numerous medications have been tried, with variable results. These are beyond the scope of this chapter, and thus will be briefly change to listed here. The main classes of treatment are chemotherapy (chlorambucil, [62] low-dose melphalan, interferon alpha, methotrexate, cyclophosphamide); immune modulators (intravenous immunoglobulin, [63] azathioprine, and systemic corticosteroids); and then few reports of other medications (dapsone, [64] clofazimine, thalidomide, [65] lenalidomide, acitretin). Medically refractory cases have also been treated with extracorpeal photopheresis with IVIG [66]. If there is an underlying paraproteinemia or lymphoproliferative disorder (i.e. multiple myeloma) then treatment has been aimed at the underlying disorder with specific chemotherapy protocols, antimyeloma protocols, or plasmapheresis.
Generalized Eruptive Histiocytoma
Clinical Features
Benign Cephalic Histiocytosis (BCH) Generalized eruptive histiocytoma (GEH) is a rare subtype of non-Langerhans cell histiocytosis that is often seen in adults, but has been reported in children as young as 1 month of age [67]. Clinically, it is characterized by asymptomatic, self-healing, recurrent crops of multiple, widespread, small tan to red-brown papules that often present symmetrically on the face, trunk, and proximal extremities. The mucous membranes are rarely involved, and the viscera are spared. Lesions appear in crops and subside spontaneously. GEH is thought to lie along a spectrum with other histiocytic disorders, and can represent the early undifferentiated stages of other non-Langerhans cell histiocytoses that share clinical and histologic characteristics, such as xanthoma disseminatum, [68] juvenile or adult xanthogranuloma, multicentric reticulohistiocytosis, and progressive nodular histiocytosis [69]. Many of these histiocytoses can have systemic involvement and can be more serious.
Investigation Recommendations
Diagnostic |
Skin biopsy for: |
Histopathology |
Immunohistochemistry |
Electron microscopy |
Diagnosis can be made by skin biopsy in conjunction with history and clinical appearance. There can be overlap between the various histiocytic conditions. Some features that may help differentiate GEH from other more serious histiocytoses are as follows:
Histology: Dense, monomorphic histiocytic infiltrate within the upper and mid dermis with a normal epidermis. Histiocytic cells have vacuolated cytoplasm. Absence of foamy histiocytes and touton cells.
Immunohistochemical stains: Negative CD1a and S100 and positive Factor XIIIa and CD68.
Electron microscopy: Reveals various nonspecific cytoplasmic organelles such as comma-shaped or worm like bodies, laminated body, dense bodies, dark lipid bodies, myeloid bodies, and popcorn or eyeball bodies. Absence of Birbeck granules.
Management Strategies
No treatment is necessary for this disorder as it is self-limited and there is no systemic involvement reported. Since there can be significant overlap with other more severe forms of non-Langerhans cell histiocytoses that can have systemic involvement, clinical follow-up is warranted to monitor for any development of systemic signs or symptoms or transformation of the disease. The course of GEH has been noted to last anywhere from 1 month to 12 years, with either complete disappearance of lesions, or residual hyperpigmented brown macules. Potential treatment options have been reported to hasten resolution and minimize recurrence.
There have been case reports in the literature of use of PUVA [70] and isotretinoin [71] in adults with GEH. PUVA therapy showed partial regression of lesions after 10 treatments, and complete resolution without recurrence after 20 treatment sessions. Isotretinoin use showed complete resolution of lesions for 8 months, but then recurrence of lesions after this period.
Progressive Nodular Histiocytoma
Clinical Features
Progressive nodular histiocytoma (PNH), also known as progressive nodular histiocytosis, is an extremely rare mucocutaneous, non-Langerhans cell histiocytosis that occurs in normolipemic individuals with no abnormalities in lipid metabolism. Onset in both childhood and adulthood have been described. PNH does not resolve spontaneously, and has an unremitting course. Clinically, it is characterized by the progressive appearance of numerous lesions of two distinct morphologies, one being widespread yellow-brown to yellow-pink papules ranging from 2 to 10 mm in size, presenting throughout the body, but sparing the flexural areas. The second morphology is large red-brown subcutaneous nodules ranging from 10 to 50 mm in size, with overlying telangiectasias often located on the trunk and/or over areas of pressure [72]. Mucosal involvement of the conjunctiva, oral mucosa, pharynx, and larynx [73] can also occur. Cutaneous lesions are benign, but can be significantly disfiguring, painful, and pruritic. Ulceration and bleeding of the papules and nodules can occur. Lesions of PNH most commonly affect the face, where they are so densely grouped that they can result in the typical appearance of leonine facies and can cause ectropion. Overall, patients are generally in good health, there is no joint involvement, and very rarely any systemic involvement [74].
Investigation Recommendations
Diagnostic |
Skin biopsy for: |
Histopathology |
Immunohistochemistry |
Electron microscopy |
Diagnosis can be made by skin biopsy in conjunction with history and clinical appearance.
Histology: Histiocytic infiltrate within the dermis with spindle-cell appearance and vacuolated clear cytoplasm, Touton-like giant cells, lymphocytes, plasma cells, and ‘collagen trapping,’ which is a result of splaying of collagen bundles at edge of the infiltrate. In older lesions there is more fibrosis present and absence of Touton-like giant cells.
Immunohistochemical staining: Negative CD1a and S100 and positive Factor XIIIa and CD68
Electron microscopy: Histiocytes with large indented nuclei and scant cytoplasm. Cytoplasm is rich with endoplasmic reticulum, Golgi bodies, lysosomes, and comma shaped bodies. Absence of Birbeck granules.
Management Strategies
The course of PNH is progressive and disfiguring. Surgical excision of large, disfiguring, or symptomatic nodules is the best therapeutic option for individual lesions, and in many cases did not show evidence of recurrence [75]. Surgical excision has been recommended in at least one article to be the only treatment option available, [76] as none of the other treatments reported in case reports have been shown to be effective.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


