Hereditary Breast Cancer: Risk Assessment, Genetic Testing, and Management Options
Beth N. Peshkin
A family history of breast cancer in first-degree relatives, especially when diagnosed premenopausally, is a strong risk factor for the development of this disease (1). Given its high incidence, particularly in Western nations, it is not uncommon for women to have a family history of breast cancer. However, the entity known as hereditary breast cancer affects only a small proportion of all women who have had such a diagnosis. Thus, the vast majority of women who have had breast cancer, or who have a family history of breast cancer, do not harbor an inherited predisposition to this disease. This chapter reviews the following topics: the genetic epidemiology of hereditary breast cancer, the identification of hereditary breast cancer based on family history collection, issues in genetic and cancer risk assessment, the role of genetic counseling and testing, and management issues in high-risk women.
Hereditary Breast Cancer Syndromes and Associated Cancer Risks
Hereditary Breast/Ovarian Cancer: Brca1 and Brca2 (Brca1/2)
Approximately 7% of breast cancer patients in the general population are believed to have an inherited basis for their disease (2). Most of these cases are attributable to mutations in the BRCA1 and BRCA2 genes, which were cloned in the mid-1990s. For example, in a study of families with four or more breast cancers and no ovarian cancers, 28% were due to BRCA1 mutations, 37% were due to BRCA2 mutations, and 35% were associated with mutations in other genes (3). However, 80% of families with multiple cases of breast and ovarian cancer were attributable to BRCA1 mutations, whereas only 15% were associated with BRCA2 mutations (3). Thus, generally speaking, in families in which there are no cases of ovarian cancer and/or fewer cases of breast cancer, the likelihood of finding a BRCA1 or BRCA2 mutation is diminished. In addition, as individuals with less suggestive family histories are tested in clinical practice, the proportion with positive BRCA1/2 results is also reduced.
The cancer risk profiles associated with mutations in BRCA1 and BRCA2 are similar, but there are some important differences. A pooled pedigree analysis from 22 studies showed that, to age 70 years, the average cumulative risk of breast cancer in women with a BRCA1 mutation is 65% (95% confidence interval [CI], 51% to 75%), and in BRCA2 carriers, it is 45% (95% CI = 33% to 54%) (4). The comparable risk in the general population is at least 7% (5). The risk of breast cancer by age 50 years is higher in BRCA1 carriers than in BRCA2 carriers, but the risk of premenopausal breast cancer is significantly elevated in both groups of carriers (4,6).
BRCA1 mutations also confer an elevated risk of ovarian cancer of 39% (95% CI = 22% to 51%), whereas the risk associated with BRCA2 mutations is lower, at 11% (95% CI = 4% to 18%) (4). The comparable risk in the general population is less than 1% to age 70 years (5). Although much of the risk in carriers occurs after age 50 years, the risk is elevated prior to that age, especially in BRCA1 carriers, in whom risk begins to rise more sharply by age 35 to 40 years (4). Note that the ovarian cancers identified in carriers are of epithelial origin, usually papillary serous adenocarcinomas (7). In addition, fallopian tube cancer is part of the BRCA1/2-associated tumor spectrum (8). Of note, oral contraceptive use reduces the risk of ovarian cancer in BRCA1/2 carriers, although it may increase the risk of breast cancer in these women (9,10).
A meta-analysis of BRCA1/2 penetrance in unaffected women based on ten studies (6) derived similar data about breast and ovarian cancer risks to those just cited (4). Furthermore, these data are available as age-specific risks, which may be useful in clinical counseling to help patients make decisions about the timing of risk-reducing surgery. For example, because the mean risk of breast cancer for a 30-year-old BRCA2 carrier is 6.6% to age 40 years, 20% to age 50 years, 35% to age 60 years, and 45% to age 70 years (6), she may feel comfortable delaying prophylactic mastectomy until age 50 years.
Mutation carriers with a prior history of breast cancer also face elevated risks of developing a second breast cancer in the contralateral breast, translating to a lifetime risk of 40% to 65% (8,11,12). In a study of 491 BRCA1/2 carriers, the risk of contralateral breast cancer at 10 years was as high as 40%; however, the risk was lower for women who were BRCA2 carriers, age 50 years or older at first breast cancer diagnosis, and who had a history of tamoxifen use and/or oophorectomy (13). For women with “sporadic” breast cancer, contralateral risks range from 0.5% to 1% per year (up to 20% at 20 years of follow-up) (14). It is unclear whether the rates of metachronous ipsilateral breast cancer are also elevated in carriers, especially in women with a history of oophorectomy (15,16,17). Because of the risk of second primary breast cancers, BRCA1/2 testing in high-risk women with a new diagnosis of breast cancer may provide valuable information to guide decisions about definitive surgical treatment (18).
The tumor spectrum in BRCA1 and BRCA2 carriers also includes male breast cancer, prostate cancer, and pancreatic cancer (8,11,19). Although the risk of prostate cancer may be as high as 40% in carriers, the risks of these other associated cancers are low overall but are still elevated relative to the general population (8,11,19).
The medical literature addressing cancer risks in BRCA1/2 carriers reveals quite a bit of variability in associated cancer risks. Aside from issues in research methodology such as ascertainment biases, other contributory factors include genetic and nongenetic modifiers of risk, the latter including reproductive
factors such as pregnancy and breast-feeding (20). Data about these factors are relatively limited but will likely be integrated into more individualized risk assessments in the future.
factors such as pregnancy and breast-feeding (20). Data about these factors are relatively limited but will likely be integrated into more individualized risk assessments in the future.
High-Risk Families Without Brca1/2 Mutations
Frequently, BRCA1/2 mutations are not identified in families in which several women have developed breast cancer. However, women in such families are still at substantially elevated risk for breast cancer (21,22). For example, Metcalfe et al. showed that in families with three or more cases of breast cancer or two or more cases diagnosed less than age 50 years in which no familial BRCA1/2 mutation is identified, women have a fourfold increase in breast cancer risk but no elevation in risk for ovarian or other cancers (23). Similarly, rates of contralateral breast cancer in affected women from BRCA1/2-negative families are also very high, approaching 40% nearly 15 years postdiagnosis (24).
Aside from BRCA1/2 mutations, other gene mutations account for less than 1% of hereditary breast cancer predisposition. The syndromes described in the following subsections are rare and tend to have characteristic features aside from breast cancer. Clinical testing is available, and a positive test result will often inform medical management. The section concludes with a description of other gene mutations that may predispose to breast cancer but for which the utility of clinical testing is unclear.
Li-Fraumeni Syndrome: Tp53
Mutations in the TP53 gene predispose to a syndrome known as Li-Fraumeni syndrome (LFS). LFS, which is inherited in an autosomal dominant fashion, is defined by a broad spectrum of early-onset malignancies, including soft-tissue sarcomas and osteosarcomas, breast cancers, brain cancers, leukemias, and adrenocortical tumors (25). Many of these cancers (except for breast cancer) are diagnosed in children. When using strict diagnostic criteria (including ascertainment through an individual diagnosed with sarcoma prior to 45 years of age), more than 50% of families are found to harbor a mutation in the TP53 gene, and a few families have mutations in the CHEK2 gene (25). The lifetime risk of cancer for women in families with LFS approaches 100%, which is largely attributed to the high risk of breast cancer (25). A preponderance of early-onset breast cancers (occurring before age 45 years) and second primary breast cancers has been reported (26). Although it is very rare for TP53 mutations to be implicated in families with early-onset breast cancers that do not exhibit other LFS-associated cancers, testing may be considered in young breast cancer patients (e.g., <30 years old) in whom BRCA1/2 mutations have been ruled out (27). In addition, concern has been raised about the potential for development of metachronous cancer within the field of radiotherapy, which may have implications for surgical treatment of women with LFS (27).
Cowden Syndrome: Pten
Cowden syndrome (CS) is a potentially underdiagnosed condition, given the broad range and sometimes subtle nature of its associated clinical features. Formal diagnostic criteria consist of specific “major” and “minor” clinical findings (28,29). The most common malignancies, which comprise three of the five major criteria, are breast, thyroid (usually follicular and never medullary), and endometrial cancer (28,29). Women with CS have a 25% to 50% risk of breast cancer, with an average age at diagnosis of 38 to 46 years (28,29). Benign breast findings, which affect up to 67% of affected women, include the presence of hamartomatous tissue, sclerosing adenosis, lobular atrophy, and fibroadenomas (28,29). Other associated findings include macrocephaly (≥97th percentile), lipomas, fibromas, hamartomatous intestinal polyps, and thyroid lesions such as goiter or follicular adenomas (28,29). Pathognomonic mucocutaneous findings include facial trichilemmomas; papillomas of the face, lips, tongue, or oral mucosa; or acral keratoses (28,29). At least 80% of patients meeting diagnostic criteria for CS have a mutation in the PTEN gene (28,29). Although these mutations are inherited in an autosomal dominant fashion, a family history of CS is often absent in affected individuals, suggesting that many cases may arise as a result of a new mutation (28,29).
Peutz-Jeghers Syndrome: Stk11
Peutz-Jeghers syndrome is a rare autosomal dominant condition characterized by hamartomatous polyps in the gastrointestinal tract, mostly commonly in the small intestine; mucocutaneous hyperpigmentation on the lips, hands, buccal mucosa, or other areas; and small bowel polyposis (30). The lifetime risk of cancer is at least 80%, which is largely attributable to markedly elevated risks of gastrointestinal malignancies (e.g., colon, stomach, and pancreas) as well as breast cancer (sometimes of early onset) and gynecologic cancers (30). When the syndrome is correctly identified, mutations in the STK11 gene are implicated in virtually all affected families (30).
Hereditary Diffuse Gastric Cancer: Cdh1 (E-Cadherin)
Hereditary diffuse gastric cancer is a rare cancer predisposition syndrome characterized by signet-ring or isolated cell–type gastric carcinoma in addition to lobular breast cancer in women (31). The lifetime risks of these cancers are greater than 67% to 83% for gastric cancer in men and women, respectively, and approximately 40% for breast cancer (31). These cancers tend to occur in individuals younger than age 40 years (31). Mutations in the CDH1 gene are found in about 30% of affected individuals (31).
Gene Mutations Conferring Intermediate and Lower Risks of Breast Cancer
Rarely occurring mutations in DNA repair genes such as CHEK2, ATM, BRIP1, RAD50, and PALB2 confer an increase in the risk of breast cancer by about twofold to fourfold (32,33). Although clinical testing is available for mutations in CHEK2 and ATM, the interpretation of a positive test result is difficult owing to imprecision in the associated cancer risks and unclear implications for risk management (32,34).
Genome-wide association studies have revealed several commonly occurring low-risk alleles, or single nucleotide polymorphisms (SNPs), that appear to contribute to breast cancer risk in a much more modest way (odds ratios between 0.88 and 1.26) (32,33,34). Depending on the number of alleles a woman has, risk appears to be multiplicative, but at most appears to be equivalent to that associated with having one or two first-degree relatives affected with breast cancer (34). Research is ongoing to identify additional SNPs and to better determine cancer risks, particularly in
the setting of a family history. Meanwhile, clinical testing for SNPs is not recommended; however, it is available through direct-to-consumer Internet sites despite the fact that the validity and clinical utility of this information remain undetermined.
the setting of a family history. Meanwhile, clinical testing for SNPs is not recommended; however, it is available through direct-to-consumer Internet sites despite the fact that the validity and clinical utility of this information remain undetermined.
Risk Assessment
Genetic Counseling
Women who are concerned about their risk of breast cancer and how to manage that risk may seek consultation from a number of providers, including gynecologists, primary care physicians, surgeons, oncologists, and genetic specialists. Because of the complexity involved in risk assessment and genetic testing, a thorough discussion of these issues often requires at least 1 to 2 hours. Thus, high-risk women and/or those who are interested in genetic testing should be referred to a genetic counselor or other genetic specialist for comprehensive genetic counseling (35). The goal of genetic counseling is to foster informed and autonomous decision making by addressing the informational, psychological, social, and familial concerns of patients. Genetic counseling is part of the process of informed consent, which is important to document prior to genetic testing (35). In the following subsections, components of and approaches to risk assessment are described.
Collection and Documentation of Family History
Recording the family history in the form of a pedigree is convenient and allows for quick inspection of patterns that may be suggestive of hereditary cancer (Fig. 21.1). Optimally, the pedigree contains information about maternal and paternal relatives over at least three generations. It is important to document all occurrences of cancer and precancerous conditions and ages at diagnosis. If possible, verification with pathology reports or medical records should be obtained, particularly for reports of “abdominal” or ovarian cancers, given that the accuracy of reported information decreases for more distant relatives (36). In addition, current ages or ages and causes of death should also be indicated for all relatives (including those without cancer). Cancer treatment and surgical history (e.g., hysterectomy and/or oophorectomy, if known), relevant exposure history, and other chronic medical conditions should be recorded. Finally, the ethnic background and country of origin of ancestors on both sides of the patient’s family should be noted.
Qualitative Features Suggestive of Hereditary Breast Cancer
Hallmark features of hereditary breast cancer include the presence of several relatives affected with breast and/or ovarian cancer, generally over two or more generations on the maternal or paternal side of the family. There is usually a predominance of early-onset cases of breast cancer (i.e., diagnosed prior to age 50 years). Bilateral breast cancer or the occurrence of breast and ovarian cancer in the same woman is also a potential indicator of hereditary cancer. Fewer cases of breast or ovarian cancer in the setting of Eastern European or Central European (Ashkenazi) Jewish ancestry may still be suggestive of an inherited predisposition, as described in the next section. The occurrence of other BRCA1/2-associated malignancies, such as prostate, pancreatic, and male breast cancer, should also be considered, as well as malignancies and conditions (e.g., sarcoma, thyroid cancer) associated with the rare syndromes described previously. Criteria for identifying high-risk individuals are given in Table 21.1 (37,38).
Individuals meeting these criteria should be referred for genetic counseling. Although the pedigree is a critical and powerful risk assessment tool, it has inherent limitations that can affect the clinician’s ability to discern a pattern of hereditary breast cancer. These include small family size, few female relatives, at-risk relatives not yet old enough to be in a high-risk age range for the development of cancer, the occurrence of oophorectomy, and limited knowledge of family medical history. Biologic considerations include variable expressivity and incomplete penetrance of the gene mutations responsible for hereditary breast cancer.
Quantitative Models of Risk Assessment
Considerations in Ashkenazi Jewish Women
The importance of ascertaining ethnic background in at-risk patients cannot be overlooked. Although hundreds of mutations
occur in the BRCA1 and BRCA2 genes, recurrent or “founder” mutations have been described in several populations, including those with French Canadian, Icelandic, Norwegian, or Dutch ancestry (39). In addition, because three recurrent mutations (i.e., 187delAG and 5385insC in BRCA1 and 6174delT in BRCA2) account for approximately 95% of those identified in Ashkenazi Jewish individuals, full testing of these genes is not routinely warranted (40,41,42). In the general population, the frequency of BRCA1 and BRCA2 mutations is estimated at 1/1,250; however, in an unselected Ashkenazi Jewish population it is 1/77 (43). Thus, it may be appropriate to consider BRCA1/2 testing in a Jewish woman with early-onset breast cancer and/or ovarian cancer and no family history. Unaffected Jewish women who have even a minimal family history of breast or ovarian cancer may also benefit from a discussion about BRCA1/2 testing.
occur in the BRCA1 and BRCA2 genes, recurrent or “founder” mutations have been described in several populations, including those with French Canadian, Icelandic, Norwegian, or Dutch ancestry (39). In addition, because three recurrent mutations (i.e., 187delAG and 5385insC in BRCA1 and 6174delT in BRCA2) account for approximately 95% of those identified in Ashkenazi Jewish individuals, full testing of these genes is not routinely warranted (40,41,42). In the general population, the frequency of BRCA1 and BRCA2 mutations is estimated at 1/1,250; however, in an unselected Ashkenazi Jewish population it is 1/77 (43). Thus, it may be appropriate to consider BRCA1/2 testing in a Jewish woman with early-onset breast cancer and/or ovarian cancer and no family history. Unaffected Jewish women who have even a minimal family history of breast or ovarian cancer may also benefit from a discussion about BRCA1/2 testing.
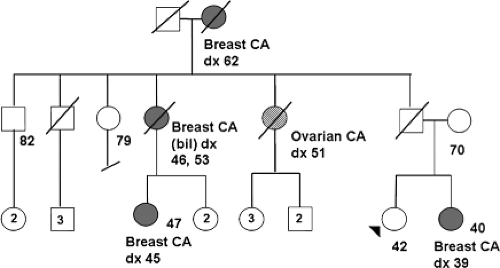 Figure 21.1. Abbreviated pedigree of a family with hereditary breast/ovarian cancer. The pedigree of this 42-year-old patient contains features consistent with hereditary breast/ovarian cancer attributable to a mutation in either BRCA1 or BRCA2. Note the presence of early onset breast cancers, bilateral (bil) breast cancer, and ovarian cancer, as well as susceptibility transmitted through a male (the patient’s father). Circles represent females; squares represent males; a line through the symbol indicates a deceased individual. Some current ages or ages at diagnosis (dx) for various types of cancer (CA) are shown. |
Table 21.1 Candidates for Genetic Counseling and Possible Testinga,b | ||
|---|---|---|
|
Assessing the Risk of Breast Cancer
For women who have not had breast cancer but who are concerned about their risk, risk assessment often begins by determining whether their family history is suggestive of hereditary breast cancer. If so, breast cancer risk may be considered in the context of the probability of carrying a mutation in a susceptibility gene such as BRCA1 or BRCA2. It may be appropriate to then discuss the possibility of genetic testing, starting with an affected relative if possible, and to refer for genetic counseling.
Women who have less extensive or suggestive family histories may be counseled about their breast cancer risk based on empiric models. Two of the most commonly utilized models, the Gail and Claus models, are available in a software program known as CancerGene (44). The Gail model is based on data from the Breast Cancer Detection and Demonstration Project (45). Subsequent modifications have been made and were used to determine eligibility for the Breast Cancer Prevention Trial (46,47). The model, which can be accessed online (48), derives age-specific risks of invasive breast cancer based on the following risk factors: age at menarche, age at first live birth, number of previous biopsies (including the presence of atypical hyperplasia), number of first-degree relatives with breast cancer, and race/ethnic background. A modification of the model that incorporates mammographic density looks promising but needs to undergo further validation; in addition, reporting of breast density must be standardized (49,50). Several caveats must be considered with this model: (a) Although the revised Gail model predicts risk for women as a group reasonably well, it has poor discriminatory accuracy for predicting the risk for an individual woman (51,52) and significantly underestimates risk in women with atypia (53). (b) The inclusion of biopsy history may falsely elevate risk, and the omission of second-degree relatives (which includes all paternal relatives) may underestimate risk. (c) This model cannot be used in women younger than age 35 years or women who do not have annual mammograms. Recently, Gail et al. reported that a similar model based on data from the Women’s Contraceptive and Reproductive Experiences study is better suited for African American women (54).
The Claus model is based on data from the Cancer and Steroid Hormone Study (2). Claus and colleagues fit various genetic models to data about age-specific risk of breast cancer among first-degree relatives of breast cancer patients and controls. The resulting model assumes that an autosomal dominant
susceptibility gene underlies the observed breast cancer rates. Risk tables are available that allow the clinician to assess absolute risk in 10-year increments based on the patient’s current age, age at breast cancer diagnosis of affected relatives, and the relationship of these relative(s) to her (i.e., first- or second-degree maternal or paternal relatives). This model is applicable only to women who have some family history of breast cancer. Other factors that may affect risk, such as those used in the Gail model, are not included in the Claus model.
susceptibility gene underlies the observed breast cancer rates. Risk tables are available that allow the clinician to assess absolute risk in 10-year increments based on the patient’s current age, age at breast cancer diagnosis of affected relatives, and the relationship of these relative(s) to her (i.e., first- or second-degree maternal or paternal relatives). This model is applicable only to women who have some family history of breast cancer. Other factors that may affect risk, such as those used in the Gail model, are not included in the Claus model.
Although the Claus model has not been subjected to validation in a population-based group of women, it has been compared with the Gail model to assess concordance. As expected, the two approaches are in agreement for certain subsets of patients and highly divergent for others (55,56). In part, these differences arise because the models are based on different assumptions and do not take into account the same parameters.
Although the Gail and Claus models consider family history in estimating breast cancer risk, some models, such as the BRCAPRO and Tyrer-Cuzick models, consider BRCA1/2 carrier probability and penetrance data as well. BRCAPRO is discussed in the following section, as it is more commonly used to assess carrier probability, particularly for women who have already had breast or ovarian cancer. However, the Tyrer-Cuzick model (also called IBIS, as it is based on data from the International Breast Cancer Intervention Study) is designed specifically and exclusively to assess breast cancer risk and gene carrier probability in unaffected women (57). The model considers individual factors such as current age, age at menarche, menopausal status, hormone use, body mass index, parity, and history of atypical hyperplasia, lobular cancer in situ (LCIS), or ovarian cancer. In addition, extensive family history of breast and ovarian cancer and prior genetic testing results are also assessed. An easy-to-use computer platform is available free of charge (58). Thus, because it incorporates multiple familial and personal risk factors, the Tyrer-Cuzick model may be especially useful in women presenting for risk assessment. In one study among 1,933 women attending a risk evaluation and screening clinic, this model proved to be the most consistently accurate in predicting breast cancer risk compared to the Gail, Claus, and BRCAPRO models (59). Within the large kConFab familial breast cancer resource, the model accurately predicted breast cancer risk in BRCA1/2-positive families more so than in mutation-negative families (60).
Assessing the Probability of Identifying A Brca1 or Brca2 Mutation
It has been suggested that a minimum estimated 10% probability of detecting a mutation is a reasonable threshold for considering testing (37,61). However, in the genetic counseling context, several models may need to be considered and interpreted within the context of qualitative pedigree analysis and patient preferences to determine the appropriateness of genetic testing (62,63). In the following, some of the most commonly used and easily applied models are briefly reviewed. In general, the presence of ovarian cancer is a significant predictor of identifying a BRCA1/2 mutation within a family, and given similar family histories, individuals of Ashkenazi Jewish descent will usually have a higher probability of testing positive than their non-Jewish counterparts. In addition, many models underpredict carrier probability in women of lower risk, thus underscoring the importance of individualized risk assessment (64).
The CancerGene program contains several commonly used models for estimating BRCA1/2 carrier probability (44). For example, the Myriad model is based on empiric observations of 10,000 consecutive gene tests performed at a commercial laboratory (40). The information is updated periodically (currently including more than 180,000 gene tests) and is downloadable to personal digital assistants (65). The data are based on the patient’s age of diagnosis of breast or ovarian cancer (if any), family history of these cancers, and the presence or absence of Ashkenazi Jewish ancestry. Limitations of these data include potential incompleteness of reported family histories and the fact that most histories were not verified by review of medical records.
Related posts:
 Follow-Up After Surgery for Primary Breast Cancer: Breast-Conserving Therapy and Mastectomy
Follow-Up After Surgery for Primary Breast Cancer: Breast-Conserving Therapy and Mastectomy
 One-Stage Immediate Breast Reconstruction with Adjustable Implants
One-Stage Immediate Breast Reconstruction with Adjustable Implants
 One-Stage Reconstruction of the Breast Using Autologous Tissue with Immediate Nipple Reconstruction
One-Stage Reconstruction of the Breast Using Autologous Tissue with Immediate Nipple Reconstruction
 Pedicled Perforator Flaps in Breast Reconstruction
Pedicled Perforator Flaps in Breast Reconstruction
 Management of Chronic Postoperative Breast Pain
Management of Chronic Postoperative Breast Pain
 The Periareolar Approach to Augmentation Mammaplasty
The Periareolar Approach to Augmentation Mammaplasty
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


