Fig. 3.1
Hyperpigmented patches in a reticulated pattern and tender nodules on the shins in a patient with polyarteritis nodosa
Traditionally, treatment of PAN aimed at inducing remission and decreasing the risk of relapse has included corticosteroids, plasma exchange, and immunosuppressant medications such as cyclophosphamide. The benefits of immunosuppressive medications must be weighed against the cost of potentially amplified hepatitis viral replication; however, since HBV therapy is more effective in the absence of PAN, it is recognized that this transient increase in viral replication during treatment for PAN is acceptable when immediately followed by interferon therapy for the HBV. Long-term survival has been shown to be highest in patients treated with a regimen of steroids, plasma exchange and cyclophosphamide rather than steroids and plasma exchange alone [3].
It is important to note that neither the severity of PAN nor prognosis in these patients is affected by the severity of the coexistent hepatitis. Instead, outcome is more closely related to the severity of the PAN itself and the status of kidney, liver, neurological, gastrointestinal system involvement [3].
Gianotti–Crosti Syndrome (Papular Acrodermatitis of Childhood)
Papular acrodermatitis of childhood or Gianotti–Crosti syndrome (GCS) is seen in children between the ages of two and six who are infected with HBV. This association is strongest in endemic regions. In the USA, however, the eruption is more strongly associated with Epstein-Barr virus infection than with HBV infection. Clinically, GCS is characterized by monomorphous lightly erythematous papules or papulovesicles in an acral distribution (cheeks, buttocks, forearms, and legs); lesions erupt over the course of 2–3 days and persist for up to 3 weeks; pruritus is variable. The rash classically spares the trunk, antecubital and popliteal surfaces, and mucous membranes. Over the course of several weeks, a fine scale may form over lesions. HBV-associated Gianotti–Crosti is usually accompanied by lymphadenopathy, HBsAg positivity, and hepatitis which peaks approximately at the time of resolution of the rash [3].
Serum-Sickness Like Reaction
A serum-sickness like syndrome is the most commonly associated skin finding in patients with acute hepatitis B infection. It occurs as a prodromal syndrome in 20–30 % of infected patients and usually resolves or improves by the time the patient develops clinical jaundice. Patients commonly present with low-grade fever, malaise, polyarthralgias, lymphadenopathy, and rash. The rash is most commonly urticarial and is often pruritic.
The pathogenesis of serum-sickness-like reaction in HBV is related to circulating antigen–antibody immune complexes comprising immunoglobulin M (IgM), HBsAg, and complement which produce the anaphylatoxins 3a and 5a responsible for the urticaria and vasculitis [3].
Patients with acute hepatitis B infection may also present with other rashes such as erythema multiforme, palpable purpura, or lichenoid dermatitis.
Dermatologic Findings in Chronic Hepatitis C Infection
HCV is a single-stranded RNA flavivirus transmitted primarily through parenteral routes via contaminated blood or blood products. The virus infects and replicates in hepatocytes and peripheral monocytes. Though six different genotypes of the HCV have been identified, types 1a and 1b account for over 75 % of cases in the USA [5]. The prevalence of chronic HCV infection worldwide is estimated at 170 million, with 3–4 million people newly infected each year [6]. The major burden of disease is related to the sequelae of chronic HCV infection which results from the majority of acute HCV cases. Consequently, hepatitis C is the most common infectious cause of chronic liver disease as well as the etiologic factor in multiple systemic disorders of other organs.
The majority of dermatologic manifestations seen with HCV are observed in cases of chronic infection. It is estimated that nearly 20 % of patients with HCV infection possess at least one skin manifestation of the disease which highlights the importance of recognition. The most commonly associated skin findings with chronic HCV include mixed cryoglobulinemia syndrome (MCS), porphyria cutanea tarda (PCT), necrolytic acral erythema (NAE), and lichen planus which are discussed in this section. Properly identifying these related skin findings in patients can have important implications on the course, treatment, and disease outcome, and in some cases may be the initial presentation of the underlying viral infection.
Mixed Cryoglobulinemia Syndrome
Cryoglobulins are immunoglobulins which precipitate in cold temperature and then dissolve in warm temperatures. Type I cryoglobulinemia is commonly associated with Waldenstrom macroglobulinemia, multiple myeloma, and chronic lymphocytic leukemia. Usually the anomalous protein in type I cryoglobulinemia is monoclonal IgM. There is no association between this type and HCV. Types II and III are considered mixed cryoglobulinemia. The term “mixed cryoglobulinemia (MC)” refers to elevated serum levels of circulating cryoglobulins composed of more than one immunoglobulin component such as rheumatoid factor which are usually IgM (rarely, IgA, IgG) monoclonal or polyclonal immunoglobulin, respectively. Ninety percent of patients with this mixed-type (II or III) cryoglobulinemic vasculitis have chronic HCV infection [2, 7]. Other causes of MC are HBV, human immunodeficiency virus (HIV) infection, or chronic liver disease.
Patients with mixed cryoglobulinemia syndrome (MCS) present with palpable purpura, weakness, and arthralgia. The palpable purpura typically involves the lower extremities or other dependent areas and may ulcerate (see Fig. 3.2). Purpura may be preceded by paresthesias or localized burning sensations [8]. Hyperpigmentation from hemosiderin deposition may replace the areas of purpura upon resolution. Patients with mixed essential cryoglobulinemia also may develop urticarial plaques and ulcerations, livedo reticularis, symmetric polyarthritis, myalgias, cutis marmorata, and fatigue. In more severe presentations, patients may also have hemorrhagic bullae. Raynaud phenomenon occurs in about one-third of MCS patients which, if severe, can lead to acrocyanosis, necrosis, and distal gangrene of the digits [2, 5, 7, 8].
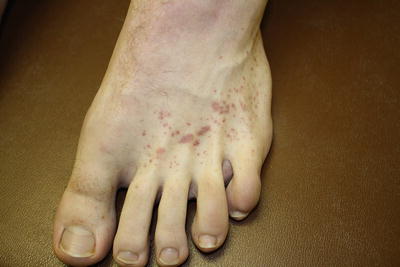
Fig. 3.2
Petechiae and palpable purpura of the feet characteristic of leukocytoclastic vasculitis associated with mixed cryoglobulinemia syndrome
Diagnosis of HCV-related MC relies on history, clinical manifestations (purpura), hypocomplementemia, and the presence of cryoglobulins. Testing for cryoglobulinemia can be difficult, as it requires serum drawn and maintained at constant temperature, before it is placed in ice bath for 30 min. 70 % of patients with MC will have positive rheumatoid factor [2]. Though not specific, positive titers for rheumatoid factor in patients with purpura and hypocomplementemia and in the absence of clinical signs of rheumatoid arthritis should raise suspicion for cryoglobulinemia [5]. Positive serology for HCV helps to confirm HCV-associated disease.
It is important to recognize that in addition to skin findings, patients with MCS often have renal disease. The renal disease associated with MC is highly variable and includes microscopic hematuria with or without chronic renal insufficiency, nephrotic syndrome, acute kidney failure, or acute glomerulonephritis. On renal biopsy, over 80 % of patients showed histologic evidence of membranoproliferative glomerulonephritis [9].
Histopathology of mixed cutaneous cryoglobulinemia reveals small vessel leukocytoclastic vasculitis, though this finding is not specific to MCS. Monocytic and lymphocytic inflammatory infiltrate in the vessel walls is usually appreciable histologically and responsible for associated endothelial hyperplasia, fibrinoid necrosis, and hemorrhage [2].
Treatment of MCS requires both immunosuppression and treatment of the underlying hepatitis. Depending on severity of disease, immune-directed modalities may include systemic corticosteroids, cyclophosphamide, rituximab, and plasmapheresis. In patients with HCV-associated MCS, immunosuppressive measures should be started at least 1 month before beginning antiviral therapy [10].
Mixed cryoglobulinemic vasculitis follows a benign course in about one half of affected patients. One-third of patients suffer subsequent liver or renal failure, and 15 % develop a malignancy, most commonly lymphoma due to the lymphotropic role of the HCV virus. While it does not integrate into the host deoxyribonucleic acid (DNA) of lymphocytes, proliferation and transformation of lymphocytes occurs by an indirect mechanism [11]. Rarely hepatocellular or thyroid malignancy follows MC and HCV. Prognosis for HCV-induced cryoglobulinemia is generally related to the response to therapy. Effective treatment for HCV can also be effective in managing the vasculitis associated with MC; however, a relapse of MC can be seen if HCV relapses after antiviral therapy. Poor prognostic factors include concomitant renal disease, which has 50 % mortality rate by 10 years of diagnosis [2].
Porphyria Cutanea Tarda
PCT is a photosensitivity disorder that results from decreased function of the enzyme uroporphyrinogen decarboxylase (UROD). Whether inherited or acquired, UROD dysfunction results in increased levels of circulating porphyrins and clinically identical PCT symptoms. HCV is a known etiologic agent in the sporadic/acquired type of PCT. In fact, up to 50 % of sporadic cases of PCT are associated with underlying HCV infection, though rates vary widely based on geography. PCT has been reported in 62–82 % of patients with HCV [5].
PCT is due to acquired or inherited depletion or absence of the UROD enzyme involved in the porphyrin pathway. HCV-related PCT is a result of diminished UROD activity in hepatocytes, In contrast to the inherited form of PCT which involves dysfunction of the same enzyme in both the hepatocyte and erythrocyte. While the exact pathogenesis of HCV-related PCT is unknown, it is likely that additional environmental factors must be present in the setting of liver disease to provoke a symptomatic PCT attack. These triggers may include alcohol, estrogens, or iron overload. Patients with chronic hepatitis and liver dysfunction have decreased intracellular glutathione stores and the resultant oxidative stresses are thought possibly to trigger PCT [5]. Increased levels of hepatocellular iron in HCV patients may trigger attacks of PCT. It has been proposed that patients who receive ribavirin therapy for HCV may develop symptomatic PCT as a result of high circulating iron levels from ribavirin-induced hemolysis [12].
Patients with PCT exhibit marked skin fragility and blisters in a photodistribution particularly on the dorsal surfaces of the extremities (commonly the hands) and the face (see Fig. 3.3). With time, vesicular lesions often become hemorrhagic before they scar and form milia. Hypertrichosis (especially of the zygomatic arches), hyperpigmentation, and sclerodermatous changes may be present in patients who exhibit a more chronic disease course [2, 13].
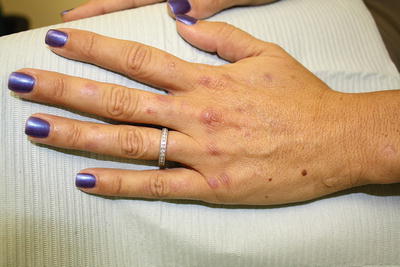
Fig. 3.3
Scarring, blisters, and milia on the dorsal hand characteristic of porphyria cutanea tarda
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


