37 Hemifacial atrophy
Synopsis





Introduction
The condition of progressive hemifacial atrophy (PHA) has no definite pathogenesis, although there have been multiple suggestions for its etiology. Most recently, Pensler et al.1 called this a lymphocytic neurovasculitis involving chronic cell-mediated vascular injury with incomplete endothelial regeneration, associated with the trigeminal nerve. Historically, it has been thought that congenital hemifacial atrophy may be a type of localized scleroderma, especially when it is noted in the forehead as the “en coup de sabre” (ECDS) or saber mark. Whether these two are different entities or whether this is the same disorder in a different form (i.e., localized scleroderma) is still not clear in the literature. In more recent years, Rogers2 reviewed 772 cases of PHA and there was a further definitive review in 1983 by Lewkonia and Lowry.3 The disorder has been shown to occur with other body asymmetries and has been described with a host of neurological features. Many neurology articles point out that, although less common, bony deformities can be present along with the soft-tissue defect. Bilateral cases have been reported but are unusual.
Historical perspective
The clinical finding of hemifacial atrophy was initially described in the writings of Dr. Caleb Hillier Parry.4 However, Parry’s medical writings were published posthumously on account of a stroke he suffered in 1816, which forced him to cease the practice of clinical medicine. With his daughter’s assistance, he continued his observations in writing, including observations about primary hemifacial atrophy. In 1825, 3 years after his death, his son Charles Parry published these writings, which included the initial description of this disorder.
In 1846, Dr. Moritz Heinrich Romberg,5 who had revolutionized the field of neurology in Europe by publishing the first systematic neurology textbook, further described the clinical manifestations of hemifacial atrophy. In 1871, Albert Eulenburg,6 a German neurologist, coined the term “progressive hemifacial atrophy” or PHA and, in 1945, Wartenberg published an extensive review article covering its clinical features.7
In the timeline of treatment options for hemifacial atrophy, it is evident that with increased availability of modern reconstructive surgical techniques the treatment of this disorder has gradually changed over the years. Blair in the 1930s,7 Sarnat and Greeley,8 and Neumann9 in the 1950s all described the use of local flaps to augment the soft-tissue deficiency. Tube pedicles were used about the same time to transfer soft tissue into the defect from remote sites with its own blood supply and for many years this was the treatment of choice. The tube pedicles were de-epithelialized with appropriate amounts of fat and dermis used for the reconstruction. In a case noted in this chapter (from the 1960s), Barsky used abdominal tissue, which was transferred to the face using this technique.
In the 1950s, bone and cartilage grafts gained favor. Campbell, and later Converse,10 described the use of onlay iliac bone graft to augment areas of atrophy. Later Longacre and De Stefano11 advocated split costal bone for the same purpose. With the advent of more biocompatible metals, Kiskadden and MacGregor described the use of tantalum for reconstruction of the hard-tissue defect.12 In the 1970s, Rees et al. published their experience of treating PHA with silicone injections.13,14 Several publications followed regarding this alloplastic approach. In the 1970s, Wells and Edgerton15 described the use of free dermis and fat from the lower abdomen as a filler material.
The era of free tissue transfer saw applications for the treatment of PHA. In the 1970s, Wallace et al. described the use of an omental free flap for soft-tissue augmentation.16 In 1985, Jurkiewicz17 similarly described the use of free vascularized tissue for patients with PHA. By the early 1990s, a series of articles appeared by Siebert and Longaker18 and others19 using free parascapular tissue for the treatment of this disorder and at the present time various modifications of this technique seem to be the standard of care.
Patient selection and treatment
• The nature and complexity of the deformity (i.e., which tissue types are affected)
• The presence of associated disorders and conditions
• The patient’s understanding of the problem and the options for treatment.
Etiopathogenesis
The exact etiology of PHA is not well understood, but is felt to have a strong autoimmune and neurogenic component. Certain histologic findings have supported a combination of the two, which may be best described as a “lymphocytic neurovasculitis.”5
Autoimmune process
PHA is likely a variant of the autoimmune disease localized scleroderma, specifically the subtype of linear scleroderma that affects the face, termed ECDS. Many times it is hard to differentiate these two entities as ECDS often leads to atrophy of subcutaneous tissue and facial bones causing hemifacial atrophy later in the disease course. Besides the clinical appearance, other etiologic, histologic, and clinical manifestations are similar between PHA and ECDS, which lends support to the theory of different spectra of the same disease. Both share similar characteristics, including age of onset, female preponderance, neurological involvement, lymphocytic infiltrate on biopsy, and a clinical course of evolution for several years followed by stabilization. Positive autoantibodies, such as antinuclear antibody, which are found in ECDS, have also been demonstrated in “classic” PHA (hemifacial atrophy without sclerodermatous skin changes).20–22
Histologic findings
The histological findings in PHA and ECDS are similar, though there are a few differences. Idiopathic PHA usually is without significant cutaneous involvement, as compared to ECDS, where cutaneous findings of hyperpigmented and/or sclerotic linear markings are expected. However, skin biopsies performed in patients with idiopathic PHA with no apparent cutaneous findings have demonstrated a cellular infiltrate similar to localized scleroderma. A perivascular infiltrate of mononuclear cells, mostly lymphocytes and monocytes, has been demonstrated in the dermis,23 with a particular focus surrounding the dermal neurovascular bundles, termed “lymphocytic neurovasculitis” by Mulliken and colleagues.1 Under electron microscopy, degenerative alterations of the vascular endothelia were also documented. These findings suggest an autoimmune disease process, similar to that of localized scleroderma.
There are a few histologic differences noted between ECDS/localized scleroderma and PHA. Although the dermal collagen fibrils appear more closely packed in patients with PHA,5 they are not as homogenized and fragmented as seen in ECDS. The elastic fibers are preserved and dermal appendages (hair follicles and sebaceous glands) are hypoplastic in PHA compared to the destruction of elastic fibers and atrophy of dermal appendages seen in ECDS/localized scleroderma.24,25
Neurogenic process
Several clinical manifestations support a neurogenic origin of PHA. The distribution of facial atrophy typically follows a dermatome of the trigeminal nerve, being unilateral in 95% of cases and only rarely crossing the midline. Pensler et al.1 reported the initial distribution of atrophy among the divisions of the trigeminal nerve in 41 patients with PHA to be 35% in V1, 45% in V2, and 20% in V3, with eventual progression of disease to involve 65% in V1, 80% in V2, and 50% in V3.5 Neuritis of the trigeminal nerve is suggested by several patients experiencing episodes of pain in the involved area prior to the onset of tissue atrophy.26 An internet survey of 205 PHA patients by Stone reported that 46% of responders experienced facial pain.27 The dermal lymphocytic infiltrate centered around neurovascular bundles in the dermis on histology also supports a neurologic target.5 Though most patients with PHA do not experience facial sensory, sympathetic, or parasympathetic dysfunction, some do experience peripheral facial nerve palsy, ocular motor palsy, and optic neuritis.28,29
Another theory involving the nervous system is the hyperactivity of the sympathetic nervous system, specifically inflammation of the superior cervical ganglion, causing features of PHA. Experimental animal studies support this hypothesis. Resende et al. ablated the superior cervical ganglion of rabbits, cats, and dogs and observed clinical features consistent with PHA within 30 days, such as localized alopecia, keratitis, enophthalmos, and hemifacial atrophy with slight bone atrophy.30 Moss et al. also observed similar findings in an experimental rat model after unilateral cervical sympathectomy.31
Clinical, radiological, and cerebrospinal fluid (CSF) laboratory findings of patients with PHA highly support that the disease affects the central nervous system (CNS), likely in an autoimmune fashion. The clinical manifestations of CNS involvement are found in approximately 8–20% of patients with PHA (the same as that found in ECDS).23 These are usually expressed as seizures, chronic headaches, and/or optic neuritis; and less commonly as neuropsychiatric disorders, deterioration of intelligence, and/or ischemic stroke. When brain imaging is performed in symptomatic patients, abnormalities such as atrophy and calcinosis are common, with up to 63% of 49 patients evaluated by Kister et al. having multiple or diffuse brain lesions on magnetic resonance imaging (MRI).28 Lumbar puncture analysis of CSF reveals findings consistent with an inflammatory process with the presence of oligoclonal bands and elevated IgG levels.30 Further evidence supporting inflammation of the CNS includes histologic findings of brain biopsies of PHA patients, which demonstrate the same changes as seen in the ECDS form of localized scleroderma: chronic perivascular lymphocytic inflammation with some vessels showing intimal thickening and hyalinization.33
Infection hypothesis
As in most autoimmune diseases, infectious agents have been postulated as an etiologic agent in PHA. The development of clinical disease manifestations has been noted to follow viral or bacterial infections. The most notorious suspect for both PHA and ECDS was Borrelia burgdorferi34,35; however, further studies have not substantiated this finding.36,37 The viral infections indicated, such as Epstein–Barr virus, correlate with the typical exposure to these infectious agents in the first and second decades of life, and are more likely coincidental rather than etiologic factors.
Trauma
The role of trauma inducing PHA is quite controversial; however, in several patients a specific history of trauma to the affected area is elucidated, especially following tooth injury or extraction.38,39 In a self-report survey of 205 patients with PHA 12% reported injuries they thought could be directly related to their disease onset.27 There have not been any standardized epidemiological studies to verify this hypothesis.
Epidemiology
The incidence of PHA is not well defined but is tightly associated with that of the ECDS subtype of localized scleroderma, since many reviews reporting and summarizing these diseases are combined together.23,27,40,41 The incidence of localized scleroderma is approximately 3 per 100 000 people and the prevalence is 50 per 100 000 people. Of those with localized scleroderma, approximately 40% have the linear subtype, and only 30% of these affect the face and/or scalp, termed ECDS.42 Therefore, an estimated incidence of 5 per 1 000 000 people and prevalence of 8 per 100 000 people is calculated for PHA. There is no racial predilection of PHA. There is a slight female predominance, with most studies having female-to-male ratios between 2.2 : 1 and 3 : 1.23,27 The median age of onset for most studies is 10 years old with a general range of 5–15 years of age,23,27,41 which is consistent with ECDS.42,43 Most cases of PHA are sporadic; however a few familial cases have been reported.44
Clinical manifestations
The initial clinical manifestations include both cutaneous findings and subcutaneous atrophy. A survey of initial symptoms reported by 49 patients with PHA demonstrates 37% having hyperpigmentation or darkening of the skin, 22% having a hypopigmented spot or streak of skin, 6% with alopecia of the scalp, medial eyebrow or eyelashes, and 24% noticed an “indentation” signifying subcutaneous atrophy (Fig. 37.1).45 The subcutaneous atrophy typically evolves first on the cheek or temple and later extends to the brow, angle of mouth, and/or neck.46 Later in the disease course, atrophy or growth arrest of the underlying bone and cartilage can occur, causing further facial deformity. Facial muscles may become atrophic, but they tend to maintain their normal function. The disease typically progresses slowly over several years (2–10 years) and then tends to enter a stable phase.38,46
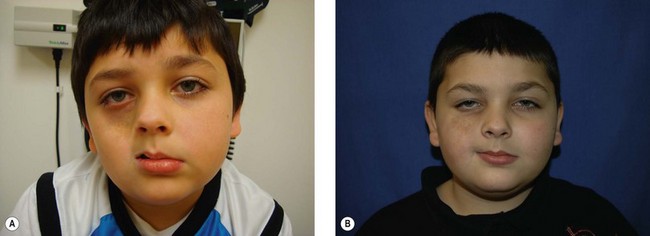
Cutaneous and subcutaneous involvement
Pigmentary changes in the cutaneous tissue are a common finding in PHA. The hyperpigmentation is often described as a “bluish” discoloration and is commonly mistakenly for a bruise that does not heal (Fig. 37.2). This is thought to reflect increased vascularity during the active or inflammatory phase of the disease.47 At times, the initial blue, violaceous, or erythematous phase is short and goes unnoticed, leaving behind a brown discoloration and/or areas of hypopigmentation. These discolorations are typically distributed in a dermatomal distribution along the trigeminal nerve.5 If the skin lesion becomes fibrotic (thickened skin) or atrophic and forms a well-demarcated linear depression (groove) in a frontoparietal or hemifacial distribution, it is considered to be ECDS morphea or linear scleroderma of the head.48
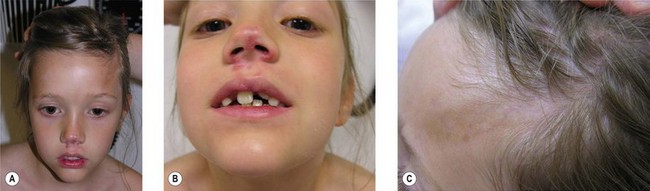
For many patients with ECDS after skin and subcutaneous induration and atrophy, the deeper tissues become involved, leading to the development of hemifacial atrophy. Therefore, the end result of ECDS appears the same as PHA without sclerodermatous changes (Fig. 37.3).

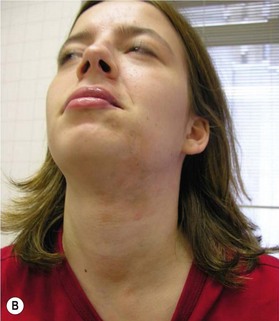
Cutaneous disease damage parameters include discoloration, both hyperpigmentation and hypopigmentation, dermal atrophy (signified by shiny skin and visible veins), subcutaneous atrophy (described as a flattening or concavity of the subcutaneous tissue), and skin thickening/fibrosis at the center of the lesion.49 Several skin appendages reside in the dermis, including sweat glands and hair follicles; therefore, alopecia of the scalp, eyebrow, and eyelashes is not uncommon.50 The severity of disease damage, in regard to subcutaneous atrophy, when evaluated by Pensler et al.1 using multivariate analysis in a group of 42 patients with PHA, was found not to be significantly influenced by trigeminal nerve distribution, side of face, age of onset, or extent (surface area) of the disease process.5
Musculoskeletal involvement
The facial musculature undergoes atrophy and thinning, mostly affecting masseteric muscles, tongue, and palatal muscles, though function is usually preserved. The degree of skeletal hypoplasia is dependent upon the age of onset, with those younger than 10 at onset having the highest risk.26






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


