Genome Instability, DNA Repair, and Cancer: Introduction
|
Introduction
The integrity of the genome of all living organisms is constantly threatened by exogenous and endogenous DNA-damaging agents. Exogenous DNA-damaging agents include physical agents, such as ultraviolet (UV) or ionizing radiation (IR), and a wide variety of chemical agents, such as components of cigarette smoke. Endogenous DNA damage arises from regular metabolic processes within the cell, mediated, for example, by reactive oxygen species. Maintaining the stability of the genome is of utmost importance to all living organisms. Therefore, since early evolution, all organisms ranging from prokaryotes to eukaryotes have been equipped with mechanisms that react to and repair DNA damage and thereby maintain genomic stability. The types of damage produced include alterations in the structure of nucleotides, DNA strand breaks, DNA cross-links, and DNA adducts. Different types of DNA-damaging agents induce different types of DNA damage (Table 110-1), which in turn require different responses and repair pathways for processing (Table 110-2).1
Agent | Damage |
|---|---|
Ultraviolet (UV) radiation | Dipyrimidine cyclobutane dimers (TT, TC, CT, or CC), pyrimidine-pyrimidone (6–4) photoproducts (mostly TC), DNA-protein cross-links |
X-irradiation | DNA single- and double-strand breaks, oxidative base damage |
Psoralens plus UVA | DNA-psoralen monoadducts, DNA interstrand cross-links (binds to T at TA sequences) |
Mitomycin C | DNA interstrand cross-links |
Benzo-[a]-pyrene | Bulky adducts |
Reactive oxygen species | Oxidative base damage (8-oxo-deoxyguanine, thymine glycol), cyclopurines (A or G) making bulky lesions |
Type of DNA Damage | DNA Repair Pathway |
|---|---|
DNA photoproducts (CPDs, 6,4-PP) | Nucleotide excision repair (NER) |
Oxidative base modifications (e.g., 8-oxoG) | Base excision repair |
Incorrect DNA base pairing | Mismatch repair |
DNA double-strand breaks | Nonhomologous end joining, homologous recombination (i.e., recombination repair) |
DNA adducts | NER |
DNA cross-links (interstrand and intrastrand) | Recombination repair |
Persistent DNA lesions | Translesion (bypass) DNA synthesis |
If DNA damage is not repaired adequately, it may lead to altered cell function, cell death, or the formation of mutations (alterations of the DNA sequence) in the damaged cells. These DNA damage-induced mutations will persist as long as the affected cell survives. At a cellular level, mutations in vital genes can lead to alterations of cell functions or malignant transformation. Accumulation of mutations may lead to organ dysfunction, aging, and cancer. Although most DNA damage is adequately repaired, none of the cellular responses is 100% effective in repairing all DNA damage under all circumstances.
A hereditary or acquired impairment in the way cells respond to DNA damage may result in genome instability with an increased rate of mutation formation. Numerous hereditary disorders are characterized by such genome instability (reviewed in Chapter 139). Many, but not all, of those are associated with an increased cancer risk and/or accelerated aging.
Exposure of the skin to UV radiation has multiple cellular and clinical effects, including an increase in skin cancer risk. The photocarcinogenesis cascade of events (Fig. 110-1) exemplifies the link between genome instability, DNA repair, and cancer. UV light produces a type of DNA damage involving the generation of photoproducts, which are alterations in the structure of nucleotides. The major DNA photoproducts are cyclobutane pyrimidine dimers (CPDs; Fig. 110-2) and 6,4-pyrimidine–pyrimidone dimers. Unrepaired CPDs or 6,4-pyrimidine–pyrimidone dimers may result in characteristic mutations: C to T single base and CC to TT tandem base substitution mutations.2–4 Such mutations are typical for UV light exposures and only rarely are induced by other mutagens. They have therefore been termed UV-signature mutations (see Fig. 110-2). Pleasance et al.5 sequenced all the genes in cells from a melanoma metastasis and reported that they harbor approximately 25,000 such mutations, accounting for about 70% of all mutations found. This clearly links the mutation burden in this cancer to prior UV-exposures.
Figure 110-1
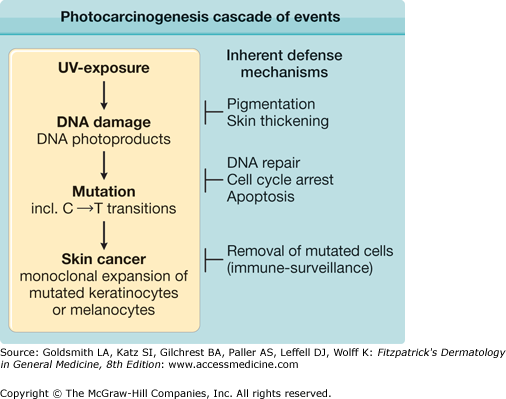
Photocarcinogenesis cascade of events. Exposure to ultraviolet (UV) light induces typical types of DNA damage, namely, cyclobutane pyrimidine and 6,4-pyrimidine–pyrimidone photoproducts. These often generate single and tandem base substitution mutations (C→T and CC→TT) that are typical for UV light exposure and are therefore termed UV-signature mutations. With sufficient numbers of inactivating mutations in crucial genes (tumor suppressor genes), individual cells may undergo malignant transformation, clonally expand, and form skin cancers. Several inherent defense mechanisms counteract this chain of events (see text).
Figure 110-2
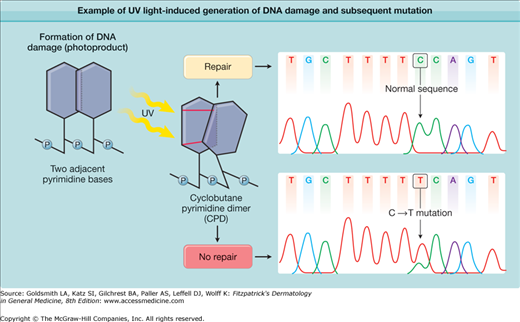
Example of ultraviolet (UV) light-induced generation of DNA damage and a subsequent mutation. On direct excitation of the DNA molecule by UV light, adjacent pyrimidine bases (cytosine or thymine) may form covalent bonds between them, which lead to creation of pyrimidine dimers. The illustration shows an example in which two covalent bindings generate a tricyclic cyclobutane ring between two pyrimidines. Hence, this type of UV light-induced DNA damage is called a cyclobutane pyrimidine dimer, a common type of DNA photoproduct. The nucleotide excision repair DNA repair system functions to remove this damage, which results in the normal DNA sequence (upper box). If the damage is not repaired, this type of DNA photoproduct can lead to formation of a typical C→T single base substitution mutation (lower box). This is most likely to occur on replication of damaged DNA and misincorporation of adenine opposite the cytosine-containing photoproduct. An example of such a UV-signature mutation is shown on the right.4 Note that the mutation is located within a run of seven pyrimidines, a common location for UV-signature mutations.
DNA repair is an important cellular defense mechanism that prevents mutation formation at sites of DNA damage after UV exposure. However, it is not the only defense mechanism (see Fig. 110-1). Most mutations are generated during replication of damaged DNA. Therefore, a damage-induced arrest in cell cycling, which allows more time for repair, is another important cellular damage response that prevents mutation formation.6,7 Furthermore, programmed cell death (apoptosis) prevents the survival of cells with overwhelming DNA damage, and through that mechanism the frequency of cells with UV-induced mutations is also reduced.8 Other inherent defense mechanisms against the ultimately carcinogenic properties of UV light include increased melanogenesis and thickening of the epidermis and stratum corneum, which protect from future DNA damage. Other protection includes removal of mutated cells through host immune responses (see Fig. 110-1).
DNA Damage and Repair
More than 100 DNA repair genes have been identified (http://sciencepark.mdanderson.org/labs/wood/DNA_Repair_Genes.html). The nucleotide excision repair (NER) pathway acts on DNA damaged by UV radiation, repairing CPDs and other photoproducts, as well as on DNA damaged by certain carcinogens (such as benzo-[a]-pyrene) (see Table 110-2). In NER, the damaged nucleotide is removed and replaced with undamaged DNA. A simplified schema of the NER system describing some of the many proteins that act in concert to repair UV-induced DNA damage is shown in Fig. 110-3. Defects in these repair genes can cause human diseases (Table 110-3), including xeroderma pigmentosum (XP), Cockayne syndrome (CS), and trichothiodystrophy (TTD) (for details on these disorders, see Chapter 139). For instance, XP can be caused by a defect in any one of several genes involved in NER. Cells/patients with defects in the same gene are considered to be in the same complementation group, and in different complementation groups if different genes are affected. The term “complementation group” is based on cell fusion experiments, in which cells from different XP patients are fused to investigate if the DNA repair defect in the fused cells is corrected. If DNA repair in the fused cell is increased, each cell provides proteins that the other is lacking and the cells “complement” each other and are in different complementation groups. If DNA repair in the fused cells is not normalized, the cells do not “complement” each other, meaning that both cells harbor mutations in the same DNA repair gene. Seven such complementation groups have been identified (XP-A to XP-G), which correspond with mutations in seven distinct genes that can cause XP.
Figure 110-3
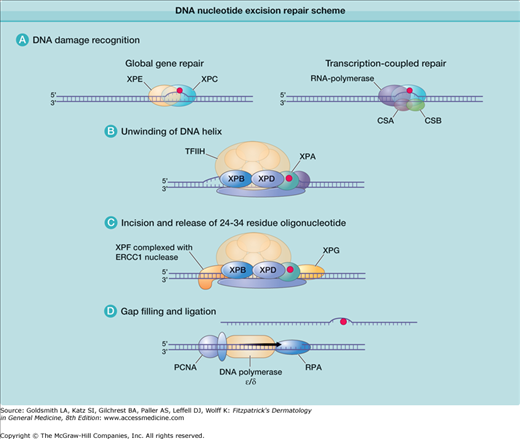
DNA nucleotide excision repair scheme. A. Right: Damaged DNA in actively transcribed genes results in stalling of the RNA polymerase in a process that involves the CSA and CSB proteins. This serves as a signal to initiate transcription-coupled DNA repair. Left: Damaged DNA in the remainder of the genome is bound by the XPE and XPC gene products. This serves as a signal to initiate global genome repair. B. A portion of the DNA, including the damage, is unwound by a complex of proteins including the XPB and XPD gene products. These proteins are also part of the 10-subunit basal transcription factor IIH (TFIIH). The XPA protein may stabilize the unwound DNA. C. The XPF and XPG nucleases make single-strand cuts on either side of the damage, releasing a 24- to 34-residue segment of DNA. D. The resulting gap is filled by DNA polymerase in a process that includes the proteins proliferating cell nuclear antigen (PCNA) and replication protein A (RPA). CSA, CSB = Cockayne syndrome complementation groups A and B; ERCC1 = excision repair cross-complementing gene 1; LIG1 = ligase 1; XPA, XPC, etc. = xeroderma pigmentosum complementation groups A, C, etc.
Geneb | Chromosome Location | Repair Pathway | Function |
|---|---|---|---|
XPA | 9q22.3 | NER | Binding of damaged DNA |
XPB (ERCC3) | 2q21 | NER | DNA helicase, part of TFIIH |
XPC | 3p25 | NER | Binding of damaged DNA, global genome repair |
XPD (ERCC2) | 19q13.3 | NER | DNA helicase, part of TFIIH |
XPE (DDB2) | 11p12-p11 | NER | Binding of damaged DNA, global genome repair |
XPF (ERCC4) | 16p13.3-p13.11 | NER | DNA endonuclease |
XPG (ERCC5) | 13q22 | NER | DNA endonuclease |
TTDA (GTF2H5) | 6p25.3 | NER | Part of TFIIH |
CSA (ERCC8) | 5q12 | NER | Transcription-coupled repair |
CSB (ERCC6) | 10q11 | NER | Transcription-coupled repair |
XPV (polymerase eta) | 6p21.1-p12 | Bypass | DNA damage bypass polymerase |
MSH2 | 2p22-p21 | MMR | Mismatch repair (Muir–Torre syndrome, HNPCC) |
MLH1 | 3p21.3 | MMR | Mismatch repair (Muir–Torre syndrome, HNPCC) |
PMS1 | 2q31-q33 | MMR | Mismatch repair (HNPCC) |
PMS2 | 7p22 | MMR | Mismatch repair (HNPCC) |
MSH6 | 2p16 | MMR | Mismatch repair (HNPCC) |
MLH3 | 14q24.3 | MMR | Mismatch repair (HNPCC) |
DKC1 | Xq28 | Telomere maintenance | Dyskerin, Posttranscriptional pseudouridylation, forms a ribonucleoprotein complex with NOP10 (gene name NOLA3) and NHP2 (gene name NOLA2) |
TERT | 5p15.33 | Telomere maintenance | Telomerase, extends nucleotide repeats at chromosome ends |
TERC | 3q21-q28 | Telomere maintenance | RNA template for telomerase |
TINF2 | 14q12 | Telomere maintenance | Part of protein complex shelterin, regulates telomere length |
NOLA2 | 5q35.3 | Telomere maintenance | See dyskerin |
NOLA3 | 15q14-q15 | Telomere maintenance | See dyskerin |
Transcribed genes are repaired faster than the rest of the genome. In the NER pathway, the first steps involving DNA damage recognition are different in nontranscribed (global genome NER) and transcribed genes (transcription-coupled NER). In nontranscribed genes and noncoding areas, which represent most of the genome, the XPE and XPC gene products bind to UV-damaged DNA, marking it for further processing. In contrast, DNA damage in transcribed genes appears to be sensed by a stalled RNA polymerase acting in conjunction with the CS complementation groups A and B (CSA and CSB) gene products. After the DNA damage recognition steps, global genome NER and transcription-coupled NER follow the same pathway. The XPA gene product functions in conjunction with replication protein A (RPA), the transcription factor IIH (TFIIH), XPF, and excision repair cross-complementing gene 1 (ERCC1). The following steps occur in both nontranscribed and transcribed gene repair:
- The XPB and XPD gene products partially unwind the DNA in the region of the damage, thereby exposing the lesion for further processing. These proteins are part of the TFIIH basal transcription factor (see Fig. 110-3B).
- The XPF gene product, in a complex with ERCC1, makes a single-strand nick at the 5′ side of the lesion, whereas the XPG gene product makes a similar nick on the 3′ side, which results in the release of a region of approximately 30 nucleotides containing the damage (see Fig. 110-3C).
- The resulting gap is filled by DNA polymerase using the other (undamaged) strand as a template in a process involving proliferating cell nuclear antigen (PCNA). DNA ligase 1 seals the region, restoring the original undamaged sequence (see Fig. 110-3D).
Regulation of Cellular Responses to DNA Damage
Several cellular responses to DNA damage contribute to the maintenance of genome integrity, including: cell cycle arrest, apoptosis (programmed cell death), and DNA repair.6,12–19 These responses need to be carefully orchestrated, and there are many proteins involved in the signaling of DNA damage and the regulation of DNA damage responses. Different types of DNA-damaging agents and different types of DNA damage require different DNA damage responses. A simplified version of this complex pathway is presented in Figure 110-4. As with defects in DNA repair genes (see Table 110-3), defects in many of these DNA damage-signaling genes (boxed in Fig. 110-4) are also implicated in hereditary disorders of genome instability (Table 110-4; for further details see Chapter 139). These disorders are characterized by an increased cancer risk, which is due to the genome instability from impaired DNA damage signaling.








