Genetic Mosaic Disorders
Lihi Atzmony
Keith A. Choate
Mosaicism refers to the presence of genetically distinct cells within an organism that result from postzygotic mutation.1 In contrast to chimerism, in which an individual is comprised of multiple cell lineages derived from distinct fertilized eggs, somatic mosaicism occurs during the development of a single embryo.2 The timing of postzygotic mutations determines the spectrum of tissues involved, with early mutations affecting pluripotent stem cells, which proliferate and migrate to affect all germ layers and give rise to widespread systemic and cutaneous disease, and with late mutations typically affecting only cutaneous progenitors and giving rise to patterned skin lesions. The patterning and appearance of cutaneous mosaic disorders is largely determined by mutation timing, its pathophysiologic effects, and by the cutaneous progenitor cells it affects.
Historically, mosaicism has been phenotypically recognized in plants, animals, and humans with manifestations including twin stripes in oranges and maze kernel, coat color variegation in animals, and skin lesions that follow the lines of Blaschko and Heterochromia Irides.2,3,4
Prior to the advent of next-generation sequencing technology, there were inadequate tools to determine the genetic basis of a mosaic disorder without prior knowledge of a candidate gene. Initial discoveries were tied to technology employed. For example, chromosomal anomalies in mosaic pigmentary disorders were initially studied via karyotyping,5,6 and more recently via sequencing and microarray-based approaches to identify chromosomal copy number variations and structural alterations. Rapid advances in understanding the molecular pathogenesis of mosaic disorders have been seen recently with paired whole exome sequencing (WES, high-throughput sequencing, next-generation sequencing) of affected and normal tissue.7,8,9
Cutaneous mosaic disorders provide a unique opportunity to study genetic mosaicism because the affected tissue is easily recognized and affected cells can be easily isolated via skin biopsy. In this chapter, we discuss phenotypic patterns and the classification of cutaneous mosaic disorders, current technologies employed to identify disease-causing mutations in mosaic disorders, and finally exemplary cutaneous mosaic disorders.
PHENOTYPE PATTERNS AND CLASSIFICATIONS
Cutaneous mosaicism can be categorized on the basis of phenotype (patterns of lesion distribution) or genetics.
Phenotype-Based Categorization
Most of the early knowledge of mosaicism was based on clinical recognition of intrapatient phenotypic variation. In 1901, Alfred Blaschko provided the first systematic documentation of cutaneous mosaic disorders. He observed more than 150 cases of various linear skin disorders such as epidermal and sebaceous nevi and carefully transposed the pattern in each patient onto a statue. A composite diagram of these distribution patterns was then published in his atlas of linear skin diseases, and these are now referred to as “Blaschko lines” (Figure 12-1).10 One hundred years later, Happle used a similar method to define more precisely the
Blaschko lines pattern of the head and neck on the basis of more than 180 figures collected from the literature.3
Blaschko lines pattern of the head and neck on the basis of more than 180 figures collected from the literature.3
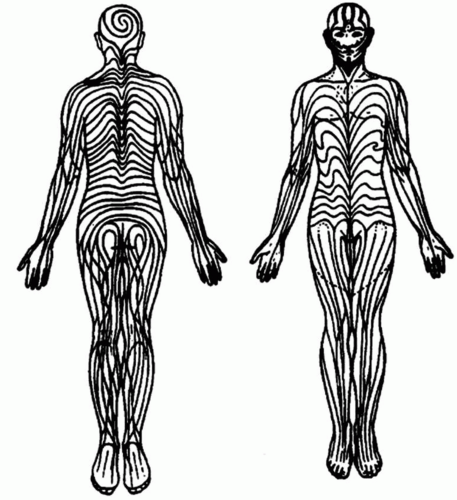 FIGURE 12-1. Lines of Blaschko. From Happle R, Mosaicism in human skin: understanding the Patterns and Mechanisms. Arch Dermatol. 1993;129 (11):1460-1470. Reprinted with permission. |
More recent classification of other patterns of cutaneous mosaicism was proposed describing five archetypical patterns of cutaneous mosaicism: Blaschko, phylloid, block-like (checkerboard), large patches without midline separation, and lateralization (Figure 12-2).11 These patterns depend on the type of cell that is affected and the patterns of migration and proliferation of its precursors during embryogenesis. Blaschko’s lines represent pathways of ectodermal development, and Blaschko-linear disorders most typically involve keratinocytes and melanocytes. Block-like and phylloid patterns respect the midline; the former is typically found in mosaicism affecting mesodermal derivatives, such as endothelial cells and fibroblasts, or neuroectodermal-derived melanocytes, and the latter is typically found in mosaicism affecting endothelial cells and melanocytes. Large patches without midline separation (coat-like pattern) are most commonly seen in highly cellular or proliferative lesions such as giant congenital melanocytic nevi or congenital hemangiomas.
More recently, a statement to classify cutaneous mosaic disorders as segmental versus nonsegmental has been proposed.12 Segmental mosaic disorders encompass the archetypical mosaic patterns that usually respect the midline. Nonsegmental mosaicism demonstrates no predictable pattern and includes single-point mosaicism, disseminated mosaicism, and patchy mosaicism without midline separation. Single-point mosaicism is the commonest type of mosaicism, with almost all solitary tumors belonging to this group. Examples include trichoepitheliomas, seborrheic keratoses, and pyogenic granulomas.12,13,14 Disseminated mosaicism encompass autosomal dominant diseases characterized by multiple tumors in which loss of heterozygosity (LOH) occurs. Examples are cylindromatosis, leiomyomatosis, tuberosclerosis, and neurofibromatosis.12
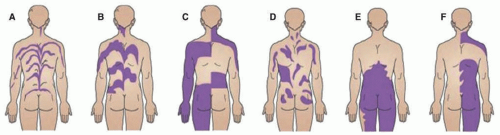 FIGURE 12-2. Patterns observed in cutaneous mosaic disorders. This includes narrow lines of Blaschko (A), broad lines of Blaschko (B), block-like or checkerboard pattern (C), phylloid pattern, (D) and patchy pattern without midline separation (E). Lateralization pattern (F) can be observed in CHILD (congenital hemidysplasia with ichthyosiform erythroderma and limb defects) syndrome, which represents functional mosaicism. Adapted with permission from Molho-Pessach V, Schaffer JV. Blaschko lines and other patterns of cutaneous mosaicism. Clin Dermatol. 2011;29(2):205-225. |
Genetic Classification of Mosaicism
Postzygotic de novo mutations initiate all forms of genomic mosaicism, which is further classified on the basis of the presence or absence of mutation in gonadal tissue. Germline (gonadal) mosaicism describes genetic heterogeneity within the gametes, permitting mutations to be inherited and expressed constitutionally by subsequent generation. An example is osteogenesis imperfecta type II. Although it was suspected that this disease is autosomal recessive, it was subsequently shown that mosaicism was common in parents of affected siblings.15 Somatic mosaicism precludes gonadal involvement, restricting mutation to somatic cells, whereas gonosomal mosaicism reflects mosaicism of both germline and somatic tissue and results from mutagenic events occurring early during embryogenesis. Although the same genetic mutation underlying autosomal dominant (AD) disorder can present in a mosaic pattern, other mutations are known to exist only in a mosaic state. As such, genomic mosaic disorders can be subdivided into two subgroups.
Mosaic Manifestation of Mendelian Disorders
These are mosaic patterns of AD disorders. In the constitutional state, AD mutations are present in all cells and are transmitted through the germline to affected offspring. Cutaneous mosaic manifestations of AD Mendelian disorders are segmental and are further subdivided into type I segmental mosaicism in which mutation is confined to the affected segment in an otherwise wild-type individual and type II segmental mosaicism in which a second mutation occurs in the subpopulation of precursor cells in an individual who already carries a germline mutation (Figure 12-3). This leads to segments with more severe phenotypes. An example of type I segmental mosaicism is segmental neurofibromatosis type I where classic skin lesions of cafe-au-lait spots and neurofibromas occur in a confined segment of the body without crossing the midline.16 Parents with segmental neurofibromatosis (NF) can produce offspring with a constitutional/generalized form of NF when gonosomal mosaicism underlies their disease.17 Examples of type II segmental mosaicism are manifestations of Hailey-Hailey and Dariers diseases with stripes of more severe disease.18,19,20
Disorders That Manifest Only as Mosaicism
These mosaic disorders are likely embryonic lethal when constitutionally expressed. Examples are listed in Table 12-1. Revertant mosaicism is a naturally occurring phenomenon involving spontaneous correction of pathogenic mutations in somatic cells. This occurs through second-site mutation that restores completely or partially the normal function of the gene. Although pathogenic somatic mutations give rise to skin lesions in cutaneous mosaic disorders, in revertant mosaicism, an island of healthy skin appears on the background of completely affected skin. Examples are ichthyosis with confetti (KRT1, KRT10) and non-Herlitz junctional epidermolysis bullosa (COL17A1, LAMB3).21,22
Epigenetic modifications are changes in gene function that are mitotically and/or meiotically heritable and that do not entail a change in DNA sequence. These include DNA methylation/demethylation, retrotransposon insertion, and imprinting.23 In skin disorders that reflect epigenetic mosaicism, a germline mutation is inherited, but its expression is modified within specific skin segments. Random X-linked inactivation (Lyonization) that occurs during early female embryogenesis can generate healthy and diseased skin, often in a Blaschkoid pattern in the setting of X-linked inherited diseases. X-linked epigenetic mosaic skin disorders can be further classified into male-lethal (eg, incontinentia pigmenti and Conradi-Hünermann-Happle syndrome), sublethal (eg, Menkes disease), and nonlethal (eg, hypohidrotic ectodermal dysplasia). In addition to Blaschkoid manifestations, other patterns of X-linked epigenetic mosaic skin disorders are lateralization seen in CHILD syndrome (congenital hemidysplasia with ichthyosiform nevus and limb defect) and block-like seen in women heterozygous for X-linked congenital hypertrichosis.24
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


