Definition and Epidemiology
Vitiligo is an acquired pigmentary dermatosis characterized by a loss of cutaneous melanocytes resulting in depigmented patches on the skin. It is relatively common, with a frequency of 1% to 2% in the United States. There is variation in the occurrence worldwide, with a frequency of nearly 9% reported in one part of India.
1 A family history is common, as is the coexistence of other autoimmune conditions. Concordance between monozygotic twins is 23%. Males and females appear to be equally affected. About 50% of patients experience onset prior to age 20, and those who experience childhood onset are more likely to have affected family members.
2,
3,
4
Etiology
Although vitiligo occurs at a high frequency in patients with a family history of the disease, it is likely a polygenic trait that results in autoimmune destruction of melanocytes. Early linkage analyses and more recent genome-wide association studies have identified a number of associations, including immune mediators, melanocyte components, and regulators of apoptosis.
5,
6 Complex interactions of these pathways, likely coupled with environmental factors, lead to the development of the clinical phenotype seen in vitiligo.
The autoimmune nature of vitiligo is supported by genetic studies as well as several key pieces of evidence: (1) There are circulating antibodies targeting melanocyte proteins.
7 (2) The clinical development of vitiligo in patients with melanoma has long been known to be a positive prognostic factor, suggesting that the antimelanocyte immune reaction evident in the skin depigmentation of these patients also targets malignant melanocytes.
8 (3) Vitiligo often coexists in patients or families with other autoimmune conditions such as Hashimoto thyroiditis and Graves’ disease, type 1 diabetes mellitus, alopecia areata, and pernicious anemia.
9,
10 (4) Finally, vitiligo is among the autoimmune phenomena that occur in patients with rheumatologic and gastrointestinal inflammatory diseases treated with tumor necrosis factor-alpha inhibitors.
11 Other hypotheses that have been put forth for the pathogenesis of vitiligo include dysregulation of the nervous system, cytotoxic mechanisms, oxidative stress-induced depigmentation, reduced melanocyte adhesion and survival, and others. Although each of these may play a role in individual patients, evidence is mounting that the underlying factor allowing or promoting melanocyte targeting is autoimmune in nature.
Clinical Presentation
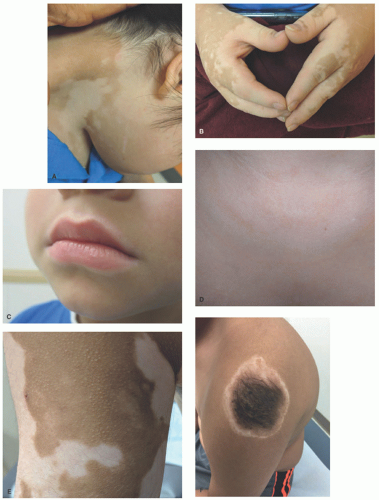
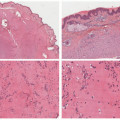
The clinical presentation of vitiligo is varied, but almost always includes depigmented, white patches and macules. Classically, it presents as symmetric, well-demarcated, depigmented patches that favor the scalp, face, neck, forearms, waist, genitalia, knees, and dorsal hands and feet (
Figure 8-1A-C). It favors areas of repetitive stress or trauma
like joints or backs of the hands and can manifest the Koebner phenomena. Widespread cases may affect any surface, and in severe cases, near-total depigmentation may occur. In patients with light skin, depigmented patches may be subtle and difficult to detect without the aid of a Wood’s lamp.
Segmental vitiligo, typically unilateral and confined to a single segment of the body with a sharp midline demarcation, shares many clinical features of nonsegmental vitiligo, but is likely predisposed by somatic mutation in a susceptibility gene, as described earlier. It is not typically associated with other autoimmune conditions, but may respond to standard therapies.
Inflammatory vitiligo may present with a raised, thread-like rim of linear erythema at the border (
Figure 8-1D), which may indicate active spread, but often remains stable for many months. Trichrome vitiligo (
Figure 8-1E), in which three distinct shades of pigmentation are visible within a body area, and vitiligo with confetti-like depigmented macules sometimes occur, and may portend a poor prognosis with rapid progression.
12 Patches of vitiligo on hair-bearing areas may present with poliosis, or depigmented, white hair. Halo nevi (
Figure 8-1F), which are melanocytic nevi with a surrounding depigmented patch, are often associated with vitiligo and are sometimes the first sign of disease.
13Spontaneous repigmentation sometimes occurs and can be hastened by a number of immunosuppressive treatment modalities. This repigmentation may be from peripheral extension (as occurs with topical steroid treatment) or may appear as perifollicular pigmented macules (as occurs with phototherapy) within the lesion from a follicular source of melanocytes.
The morbidity of vitiligo is primarily evident in its impact on the quality of life. Affected children and adults often suffer from significant psychosocial stress, depression, and anxiety.
14,
15
Histologic Findings
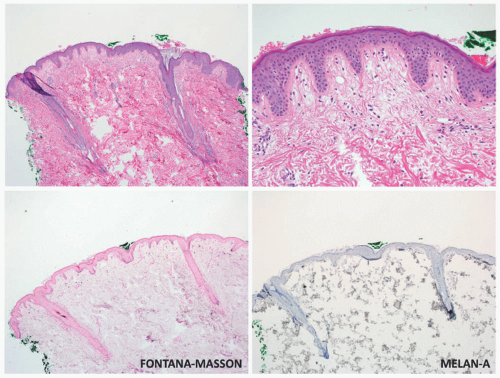
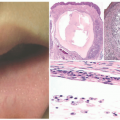
Although vitiligo is primarily a clinical diagnosis, biopsy may sometimes be necessary to differentiate it from other diseases of hypo-or depigmentation. At scanning magnification, biopsy specimens may appear completely unremarkable. Well-developed lesions are characterized by a complete absence of melanocytes and pigment (
Figure 8-2). The absence of pigment may be confirmed by negative
Fontana-Masson staining, and a lack of melanocytes may be evident by immunohistochemical studies. Inflammation is sometimes present, both in specimens that appear clinically inflammatory and in those that do not, in the form of a mild superficial lymphohistiocytic infiltrate. A more exuberant, lichenoid interface dermatitis is sometimes seen, and can appear in clinically inflamed areas, depigmented areas, and even distant, clinically uninvolved skin.
16
Differential Diagnosis
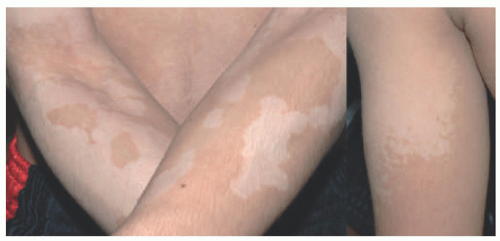
There is a broad clinical differential diagnosis for vitiligo. Hypopigmented lesions may sometimes be difficult to differentiate from depigmented lesions, and can be present in many conditions, including nevus depigmentosus/pigmentary mosaicism, pityriasis alba, and tinea versicolor (
Figure 8-3). Postinflammatory hypo-and depigmentation should be preceded by inflammation, although a history of skin inflammation is not always elicited. Chemical leukoderma may present with a strikingly similar pattern to that of vitiligo and is often occupational in nature.
Conditions of congenital depigmentation, such as Waardenburg syndrome or piebaldism, should be considered for midline patches affecting the face and trunk. These patches may sometimes be present on the extremities as well. Syndromic vitiligo may be seen in Vogt-Koyanagi-Harada, Alezzandrini syndrome, and autoimmune polyendocrinopathy candidiasis-ectodermal dystrophy syndromes.
The histologic differential diagnosis includes chemical leukoderma, piebaldism, and nevus depigmentosus. These may be indistinguishable from vitiligo and require clinical history to differentiate. In inflammatory lesions, vitiligo may mimic lichenoid dermatoses such as lichen planus and its subtypes. These conditions lack clinical depigmentation, although hypopigmented lesions may appear similar to early lesions of vitiligo. Finally, patch-stage mycosis fungoides (MF) and inflammatory lesions of vitiligo may share clinical and histopathologic findings. MF, however, should have some degree of lymphocytic atypia, and Pautrier microabscesses may be present. In some cases, multiple biopsies may be necessary to distinguish the two entities.