TABLE 7-1. Selected nonsyndromic ichthyoses | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
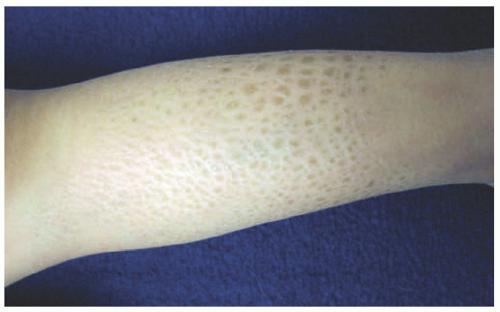 FIGURE 7-1. A child with ichthyosis vulgaris. The lower leg shows dry, polygonal, hyperpigmented scaling. |
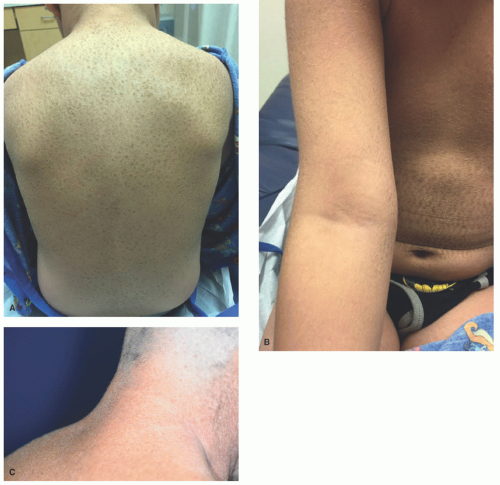 FIGURE 7-2. X-linked recessive ichthyosis with characteristic hyperpigmented scale on the back extending onto the neck (A and B) and conspicuous sparing of the antecubital fossa (C). |
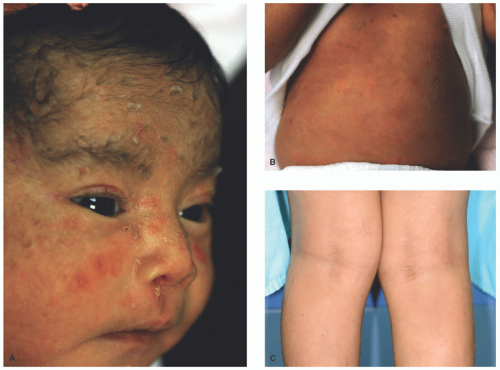 FIGURE 7-3. Epidermolytic ichthyosis. A baby showing blisters in the face, nose (A), and trunk (B). Progressive corrugated hyperkeratosis is seen in olden children, in association with a pattern of accentuation in flexural sites (C). |
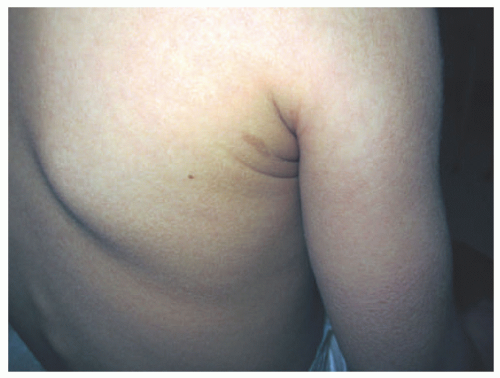 FIGURE 7-4. Autosomal recessive congenital ichthyosis with a congenital ichthyosiform erythroderma phenotype, showing fine, diffuse scale with mild underlying erythema. |
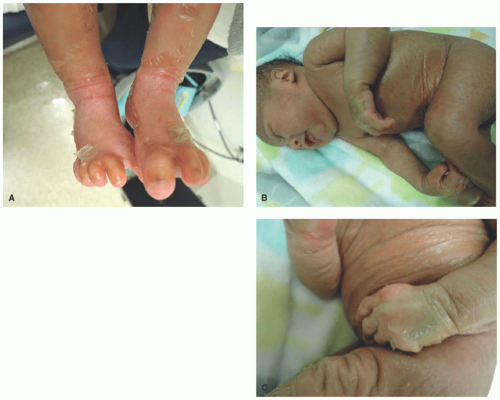 FIGURE 7-5. Collodion baby. This neonate is encased in a thick, yellowish, taut, glistening, parchment-like membrane that restricts its movements (A-C). |
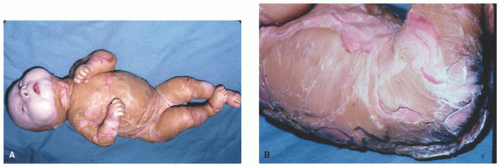 FIGURE 7-6. An infant with Harlequin ichthyosis with extremely thick, fissured scaled skin (A and B). Courtesy of Dr Kenneth Greer. Department of Dermatology. University of Virginia. |
TABLE 7-2. Histopathologic findings for selected nonsyndromic ichthyoses | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
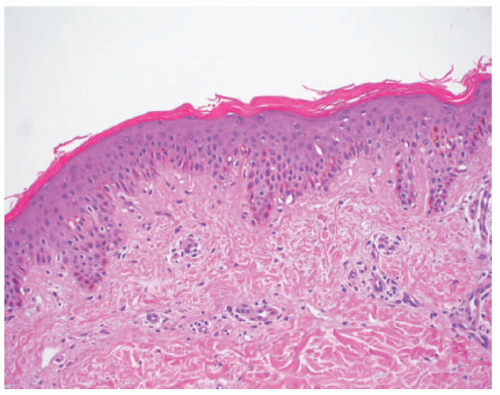 FIGURE 7-7. Ichthyosis vulgaris. Compact orthokeratosis and a diminished granular layer. |
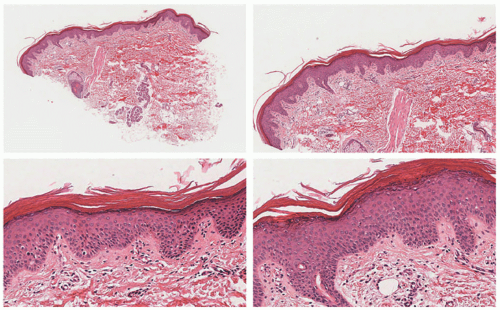 FIGURE 7-8. X-linked recessive ichthyosis. Mild epidermal acanthosis and hyperorthokeratosis. The granular cell layer is preserved (as opposed to ichthyosis vulgaris). There is mild hyperkeratosis around the sweat gland orifices. Digital slides courtesy of Path Presenter.com. |
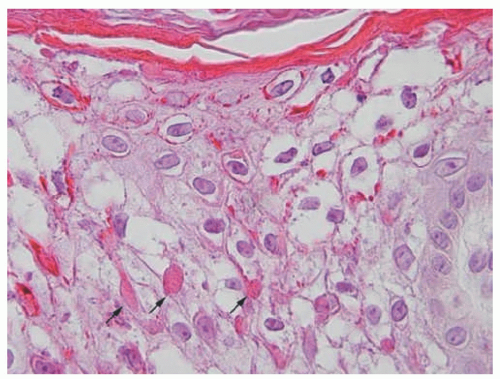 FIGURE 7-9. Epidermolytic hyperkeratosis with cytolysis of keratinocytes in the upper spinous layer and eosinophilic inclusions. Reprinted with permission from Bergman R, Khamaysi Z, Sprecher E. A Unique pattern of dyskeratosis characterizes epidermolytic hyperkeratosis and epidermolytic palmoplantar keratoderma. Am J Dermatopathol. 2008;30(2):101-105. |
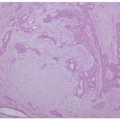
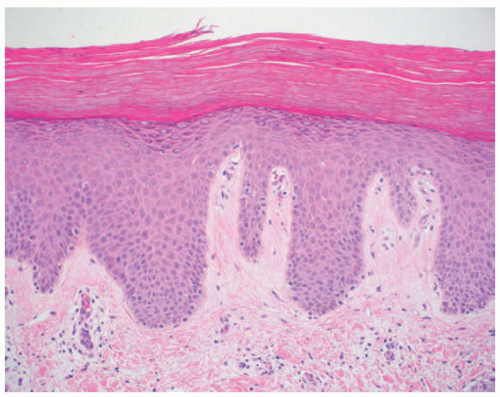 FIGURE 7-10. Lamellar ichthyosis. Thick hyperkeratosis with acanthosis and mild hypergranulosis. |
acanthosis. Focal parakeratosis, hypogranulosis, and exocytosis of neutrophils are variably present (Figure 7-14).47,48,49 In erythrodermic cases, parakeratosis can be confluent. A perivascular lymphohistiocytic infiltrate may be present in the superficial dermis.50 Periodic acid-Schiff staining demonstrates densely staining granules in upper epidermal layers.8 A microscopic evaluation of affected hair shafts reveals trichorrhexis invaginata, with a telescoping of the distal hair shaft into the proximal shaft.43 Other hair shaft findings, such as trichorrhexis nodosa, pili torti, and “matchstick” abnormality, can also be seen.
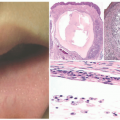
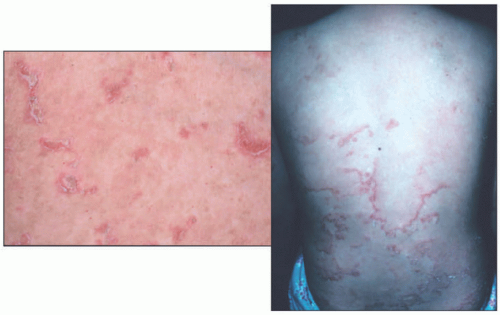 FIGURE 7-11. Ichthyosis linearis circumflexa in Netherton syndrome with the classic circinate, scaly, erythematous plaques with a border of double-edged scale. Courtesy of Dr Kenneth Greer. Department of Dermatology. University of Virginia. |
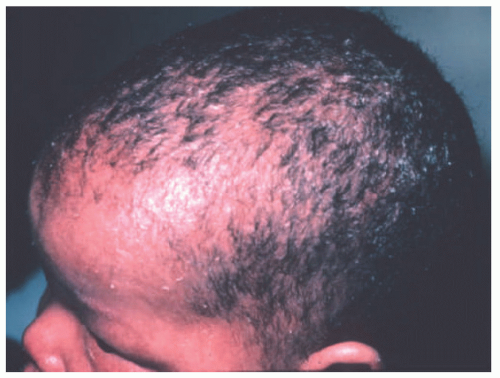 FIGURE 7-12. Congenital ichthyosiform erythroderma-like scale in Netherton syndrome. Diffuse scalp erythema and mild thin scaling is present. Courtesy of Dr Kenneth Greer. Department of Dermatology. University of Virginia. |
 FIGURE 7-13. Trichorrhexis invaginata (Bamboo hair), typical of Netherton syndrome. Courtesy of Dr Kenneth Greer. Department of Dermatology. University of Virginia. |
syndromic and nonsyndromic ichthyoses, eczematous dermatitis, and dermatophytosis.
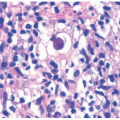
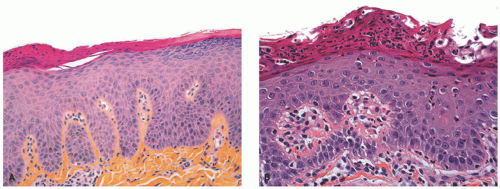 FIGURE 7-14. Skin biopsies from Netherton syndrome show psoriasiform hyperplasia and may have overlying parakeratosis (A) and neutrophils within the epidermis and stratum corneum (B). Differentiation from psoriasis must be made on a clinical basis. Reprinted with permission from Leclerc-Mercier S, Bodemer C, Furio L, et al. Skin biopsy in Netherton syndrome: a histological review of a large series and new findings. Am J Dermatopathol. 2016;38(2):83-91. |
progressive neurologic abnormalities and mild ichthyosis. Symptoms and progression are mitigated by a diet low in phytols. Skin biopsy specimens show hyperorthokeratosis and a diminished granular layer and can be differentiated from ichthyosis vulgaris by the presence of lipid accumulation on electron microscopy and by clinical history.
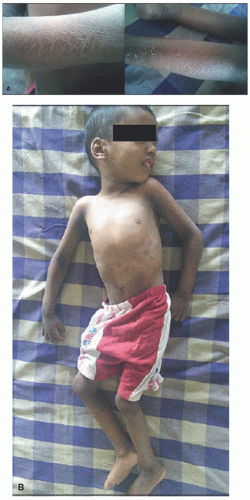 FIGURE 7-15. Marked ichthyosis in a patient with Sjögren-Larsson syndrome. A, Brown color diamond-shaped adherent scales on the upper limb and peeling of skin on the lower limb. B, Kyphoscoliosis of the trunk is present. Obtained with permission from Subramanian V, Hariharan P, Balaji J. Sjögren-Larsson syndrome: a rare neurocutaneous disorder. J Pediatr Neurosci. 2016;11(1):68-70, Figure 1 and 2. |
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


