Hormone or binding protein
Younger adults
Older adults
Men
Reference
Women (premenopause)
Reference
Men
Reference
Women (postmenopause)
Reference
DHEAS
≈12 μmol/L
Labrie et al. [2]
≈ 6 μmol/L
Labrie et al. [2]
≈3 μmol/L
Labrie et al. [2]
≈ 1.5 μmol/L
Labrie et al. [2]
Age 20 years
Age 20 years
Age 60 years
Age 60 years
8.5 μmol/L
Ravaglia et al. [3]
7.7 μmol/L
Ravaglia et al. [3]
1.5 μmol/L
Ravaglia et al. [3]
1.0 μmol/L
Ravaglia et al. [3]
(3110 ng/mL) Age
(2,824 ng/mL)
(551 ng/mL)
(364 ng/mL)
Age 90–99
<40 years
Age < 40 years
Age 90–99
Age 90–99
7.0 ± 4.0 μmol/L
Feldman et al. [4]
3.64 μmol/L
Kaaks et al. [5]
4.3 ± 2.7 μmol/L
Feldman et al. [4]
1.9 nmol/L
Burger et al. [6]
(2,600 ± 1,500 ng/mL)
(95 % CI 1.6–1.72)
(1,600 ± 1,000 ng/mL)
(95 % CI 0.8–4.4)
Ages 40–70
Ages 46–80
Median age, 54
DHEA
20–25 nmol/L
Burger [7]
20–27 nmol/L
Burger [7]
4–6 nmol/L
Labrie et al. [8]
6–7 nmol/L
Labrie et al. [8]
Ages 20–30
Ages 20–30
Ages 50–80
Total testosterone
13–40 nmol/L
Harman et al. [9]
0.6–2.5 nmol/L
Burger [7]
6–26 nmol/L
Harman et al. [9]
0.56 ± 0.26 nmol/L
Laughlin et al. [10]
Age 30
(0.2–0.7 ng/mL)
Age 80
Ages 50–89
Age unspecified
18.0 ± 6.1 nmol/L
Feldman et al. [4]
1.66 nmol/L
Kaaks et al.[5]
15.7 ± 5.6 nmol/L
Feldman et al. [4]
1.4 nmol/L
Burger et al.[6]
(5.2 ± 1.8 ng/mL)
(95 % CI 1.6–1.72)
(4.5 ± 1.6 ng/mL)
(95 % CI 0.6–2.4)
Ages 40–70
Age < 50
Ages 46–80
Median age, 54
22 ± 7 nmol/L
Gapstur et al. [11]
1.2 ± 0.7 nmol/L
Sinha-Hikim et al. [12]
15.8 ± 0.17 nmol/L
Labrie et al. [13]
0.5 ± 0.01 nmol/L
Labrie et al. [13]
(4.57 ± 0.05 ng/mL)
(0.14 ± 0.004 ng/mL)
Ages 69–80
Ages 55–65
Free testosterone
0.097 ± 0.039 ng/mL
Feldman et al. [4]
12.8 ± 5.5 pmol/L
Sinha-Hikim et al. [12]
0.075 ± 0.032 ng/mL
Feldman et al. [4]
0.14 ± 0.07 nmol/L
Laughlin et al. [10]
(0.34 ± 0.14 nmol/L)
(0.26 ± 0.11 nmol/L)
Ages 50–89
Ages 40–70
Ages 46–80
DHT
0.91 ± 0.58 nmol/L
Feldman et al. [4]
0.43 nmol/L
Secreto et al. [14]
1.19 ± 0.70 nmol/L
Feldman et al. [4]
0.22 ± 0.11 nmol/L
Secreto et al. [15]
(0.26 ± 0.17 ng/mL)
(0.12 ng/mL)
(0.35 ± 0.20 ng/mL)
(62.4 ± 32 pg/mL)
Ages 40–70
Ages 30–49
Age ≤69
SHBG
32 ± 16 nmol/L
Feldman et al. [4]
53.1 nmol/L
Kaaks et al. [5]
32 ± 16 nmol/L
Feldman et al. [4]
50.5 nmol/L
Burger et al. [6]
Ages 40–70
(95 % CI 50.7–50.5)
Ages 46–80
(95 % CI 21.0–99.8)
Age <50
Median age, 54
Estradiol
84.0 ± 22.4 pmol/L
Vermeulen et al. [16]
381 pmol/L
Kaaks et al. [5]
88.1 ± 24.6 pmol/L
Vermeulen et al. [16]
18 ± 9 pmol/L
Laughlin et al. [10]
Ages 24–31
(95 % CI 356–407.6)
Ages 70–79
Ages 50–89
81.5 ± 23.1 pmol/L
Vermeulen et al. [16]
79.0 ± 1.1 pmol/L
Labrie et al. [13]
15.4 ± 0.7 pmol/L
Labrie et al. [13]
Ages 37–46
(21.5 ± 0.3 pg/mL)
(4.2 ± 0.2 pg/mL)
Ages 69–80
Ages 55–65
Estrone
156 ± 75 pmol/L
Feldman et al. [4]
409.9 pmol/L
Kaaks et al. [5]
124 ± 78 pmol/L
Feldman et al. [4]
70 ± 34 pmol/L
Labrie et al. [13]
Ages 40–70
(95 % CI 383.4–436.4)
Ages 46–80
Ages 50–89
139 ± 2 pmol/L
Labrie et al. [13]
65.9 ± 1.8 pmol/L
Labrie et al. [13]
Ages 69–80
Ages 55–65
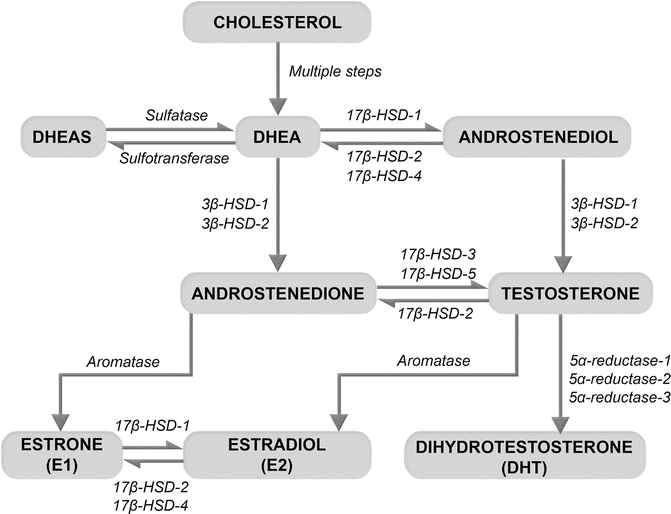
Fig. 11.1
Production of sex steroids from the adrenal precursors, dehydroepiandrosterone (DHEA) and its sulfate form (DHEAS). DHEA, an inactive prohormone, is produced by the adrenal glands from cholesterol. DHEA and DHEAS are interconvertible by the actions of sulfatases and sulfotransferases. In peripheral tissues, the hydroxysteroid dehydrogenases (HSD) convert DHEA to androstenedione and androstenediol, precursors of testosterone. The action of 5α-reductases converts testosterone to its potent metabolite, dihydrotestosterone (DHT). Aromatases produce estrogens by converting testosterone to estradiol and androstenedione to estrone
The action of sex steroids on target tissue depends both on circulating levels and on local formation within the tissue itself (Fig. 11.2). Androgens and estrogens produced within tissues can act on neighboring cells (paracrine activity) or within the same cells (intracrine activity). Sex steroids produced in peripheral tissues (such as adipose tissue) also enter the circulation, and this source becomes especially significant as gonadal production declines with age. In postmenopausal women, for example, peripherally synthesized estrone is the primary source of estrogen. In men aged 60–75, adrenal DHEA contributes about 40 % to the total pool of androgens [18]. In older men and women, comparable amounts of sex steroids are synthesized outside the gonads [8, 18]: using circulating DHT metabolites as a measure, it is estimated that postmenopausal women synthesize almost half as much androgen as men of similar age, the excess in men being attributable to testicular origin [13].
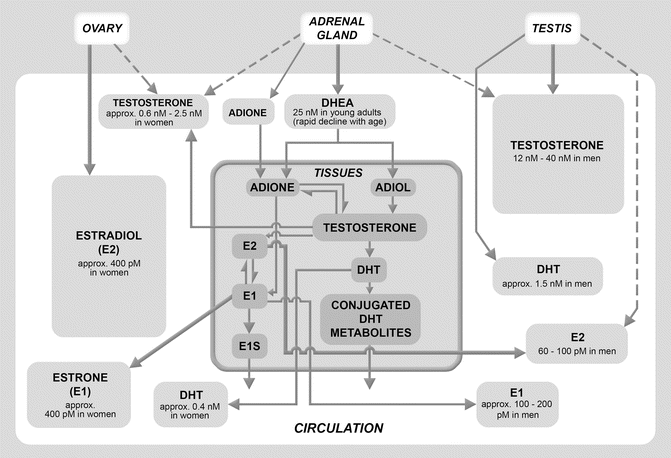
Fig. 11.2
Sources of the sex steroids in young men and women. Sex hormones are produced by the gonads, the adrenals, and by peripheral tissues. The ovary, adrenal gland, and testis are shown at the top of the chart. The white box in the central field denotes the circulatory compartment. The left side of the circulatory compartment below the ovary shows circulating levels of the sex hormones in young women; the right side of the circulatory compartment, below the testis, shows circulating levels of sex hormones in young men. The gray box with a bold border below the adrenal gland denotes the peripheral tissue compartment and illustrates peripheral conversion of the adrenal prohormone, DHEA, to sex hormones. Bold arrows denote primary sources of the sex hormones. The ovaries and testes are primary sources of circulating estradiol and testosterone in young women and men, respectively. The peripheral tissues (mainly adipose tissue) are a primary source of circulating estradiol in men and of estrone in both sexes. DHEA dehydroepiandrosterone, ADIONE androstenedione, ADIOL androstenediol, DHT dihydrotestosterone, E2 estradiol, E1 estrone
11.3 Age-Related Changes in the Sex Steroids
Overall trends in circulating levels of sex hormones in aging men and women are summarized in Table 11.2. Factors affecting circulating levels of prohormones, androgens, and estrogens in each sex are described below.
Table 11.2
Trends in circulating levels of sex steroids in aging men and women
Prohormone or hormone | Change with advancing age | |
|---|---|---|
Men | Women | |
DHEAS | Substantial decrease | Substantial decrease |
DHEA | Decrease | Decrease |
Testosterone | Decrease | Decrease |
DHT | No significant change | No significant change |
Estradiol | No significant change | Substantial decrease |
Estrone | No significant change | Decrease |
11.3.1 Changes in Prohormone and Androgen Production
Production of DHEA and DHEAS begins during adrenarche. Serum concentrations of DHEAS reach their peak (in the order of 10−8 and 10−6 M, respectively) between the ages of 20 and 30 years [8]. Then DHEAS concentrations decline with age, being reduced to 20 % of peak levels by age 70 and 5 % of peak levels by age 90 [4, 6, 19–21]. Because DHEAS and DHEA serve as sex steroid precursors in peripheral tissue, this decline in circulating levels is thought to contribute to some of the degenerative changes seen with aging.
Gender differences exist in serum concentrations of these hormones (see Table 11.1). Circulating levels of DHEAS in adult women are consistently lower than those in men at all ages [20, 22, 23]. The decline in these prohormones is clinically important in both sexes, but especially so in women. In men, gonadal androgen production declines slowly, such that peripheral production of contributes some 40 % to the total androgen pool in men by age 65 [18]. By contrast, in postmenopausal women, DHEA is the exclusive source of sex steroids for all tissues except the uterus [18]. Nevertheless, large variability exists in the circulating levels of DHEA among women: an almost eightfold difference between high and low levels has been found [24], with the low end being barely detectable. This wide range could help explain why some older women experience fewer signs of hormone deficiency after menopause, while others experience significant signs and symptoms [24].
About 30–50 % of total androgens in adult men [25] and about 50–100 % in adult women, depending on age, are derived from DHEA and DHEAS [26]. In women, androstenedione levels decline with age up to menopause, remain fairly stable in the early years after menopause [10, 27], and then decline by about 20 % by 30 years after menopause [10]. Early studies suggested that the postmenopausal ovary was a source of androgens [26], but this is controversial. Recent studies indicate that expression of steroidogenic enzymes by the postmenopausal ovary is limited [28, 29], and that, absent the adrenal production, postmenopausal women have no detectable circulating androgens [30].
Testosterone and its highly potent metabolite, dihydrotestosterone (DHT), exert receptor-mediated activity. About 1–2 % of circulating testosterone is free, 32 % loosely bound to albumin, and 66 % bound to sex hormone-binding globulin (SHBG). Free and albumin-bound testosterones are bioavailable to tissues.
In younger men, the testicular Leydig cells produce 95 % of testosterone and a smaller amount of DHT (see Fig. 11.2). In premenopausal women, 25 % of circulating testosterone comes from the adrenals, 25 % from the ovaries [31], and the rest from peripheral conversion of androstenedione in adipose tissue [32].
In healthy men, total testosterone and free testosterone decline slowly with age [4, 33, 34]. The decline in total testosterone is modest, but comparatively greater for free testosterone due to the concurrent age-related rise in SHBG [35]. Data from a large, population-based cohort of men aged 40–70 at baseline who were followed for 7–10 years (the Massachusetts Male Aging Study) showed that total testosterone declined by age cross-sectionally (between subjects) at 0.8 %/year, free and albumin-bound testosterone declined at about 2 %/year, and SHBG concentrations rose about 1.6 %/year. Longitudinally (within subjects), total testosterone declined 1.6 %/year, bioavailable testosterone declined by 2–3 %/year, and SHBG levels rose at 1.3 %/year [4].
The age-related decline in testosterone in men is due to reductions in the number of productive Leydig cells as well as to changes in the response to leutinizing hormone (LH) and human chorionic gonadotropin (hCG). Moreover, obesity is associated with lower total and free testosterone and SHBG; a change in BMI from nonobese (<25 kg/m2) to obese (≥30 kg/m2) is equivalent to a 15-year fall in testosterone levels [35]. It has been postulated that age primarily affects testicular function, whereas obesity impairs hypothalamic and pituitary function. In addition, the circadian rhythm, with higher testosterone levels in the morning than in the evening, is lost in older men [36].
In women, testosterone levels decline between the ages of 20 and 50 [37]. However, the menopausal transition itself does not appear to affect testosterone levels significantly. A prospective study in 172 women failed to show a change in total testosterone in the time span from 5 years before to 7 years after the final menstrual period [6]. Other studies found a slight decrease in levels of bioavailable testosterone in the early years after menopause [10, 27], followed by a rise to premenopausal levels in the second decade following the menopausal transition [10]. Hence, declines during the decades preceding menopause have the most significant impact on testosterone levels in older women. These observations may reflect the relative importance of adrenal and peripheral sources of androgens in women.
11.3.2 Changes in Estrogen Production
In women of reproductive age, the ovaries are the principal source of estradiol. Menopause occurs when senescence of the ovarian follicles reduces the gonadal production of estradiol to miniscule levels (see Table 11.1). During this transition, circulating levels of estradiol decline from over 300 pmol/L to about 20 pmol/L [27, 38].
Estrone, a weaker estrogen produced from androstenedione in peripheral tissues, is the predominant form of estrogen in postmenopausal women (see Fig. 11.2). Estrone is also the principal source of postmenopausal estradiol, although only 5 % of estrone is thus converted [39]. Adipose tissue is a major site of peripheral estrone synthesis; hence, estrogen levels are higher in postmenopausal women with a high body mass index.
In men, 80 % of plasma estradiol is produced by peripheral aromatization of testosterone, which occurs principally in fat tissue [40]; the testes produce only 20 % of plasma estradiol [41, 42]. In younger men, 20 μg/day of estradiol derives from peripheral conversion of plasma testosterone, 5 μg/day from androstenedione, and 5–10 μg/day from the testicular Leydig cells [16]. As in women, plasma estrone derives principally from tissue aromatization of androstenedione, with 20 % secreted directly by the adrenals. The mean plasma estradiol concentration in men is 2–3 ng/dL (about 80 pmol/L), and the mean concentration of estrone is 3–6 ng/dL (about 100–200 pmol/L) (see Table 11.1) [43].
In men, plasma estradiol levels do not decrease significantly with age [19, 33, 44] (see Tables 11.1 and 11.2), although some investigators have reported declines [45, 46]. It has been postulated that plasma estradiol levels remain relatively unchanged despite declines in testosterone because of a rise in aromatase activity coincident with the age-associated increase in body fat mass [47, 48]. Consequently, plasma estradiol levels in older men are significantly higher than in postmenopausal women (see Table 11.1).
11.3.3 Intracrine Production of the Sex Steroids
The skin is affected not only by the action of circulating sex steroids but also by locally synthesized androgens and estrogens. Local production depends on the expression of androgen- and estrogen-synthesizing enzymes in individual skin structures and cell types (Table 11.3). Moreover, the expression of sex steroid receptors in the skin varies by cell type and by sex. Cytochrome p450c17, an enzyme necessary for the synthesis of DHEA and androstenedione from cholesterol, is not expressed to a great extent in androgen target cells, such as sebocytes; however, sebocytes, sweat glands, and dermal papilla hair cells do express enzymes that convert these adrenal prohormones into biologically active testosterone and DHT [49, 50]. Indeed, human sebocytes selectively use DHEA to produce active androgens [51]. A newly identified pathway in human sebocytes synthesizes DHT from DHEA without requiring testosterone as an intermediate. The sebaceous glands, the outer and inner root sheath cells of anagen terminal hair follicles, and dermal papilla cells express aromatases that convert testosterone and androstenedione into estrogens (see Table 11.3) [52, 53].
Table 11.3
Localization of sex steroidogenic enzymes and androgen and estrogen receptor activity in the skin
Skin structure | Enzyme activity | Sex steroid receptors | |||||
|---|---|---|---|---|---|---|---|
17β-HSD | 3β-HSD | 5α-Reductase | Aromatase | AR | ERβ | ERα | |
Epidermal keratinocytes | + (4- isotype) | + (1- isotype) | + | + | |||
Melanocytes | + | + | + | ||||
Hair follicles | + (1- isotype) (2- isotype in beard) | ||||||
Follicular keratinocytes | + | + | |||||
Root sheath | + (2-isotype) | + | + | ||||
Matrix epithelium | + | + | |||||
Dermal papilla cells | + | + | + | ||||
Sebaceous glands | + (3- and 5- isotypes) | + (1- isotype) | + (1- isotype) | + | + | + | + |
Sweat glands | + (1- isotype) | + | |||||
Eccrine | + | ||||||
Apocrine | + | + | + | ||||
Dermal fibroblasts | + | + | + | ||||
Endothelial cells | + | + | |||||
As in other steroidogenic organs, six enzyme systems are involved in the activation and deactivation of androgens in the skin: steroid sulfatase, 3β-hydroxysteroid dehydrogenase Δ5−4 isomerase (3β-HSD), 17β-hydroxysteroid dehydrogenase (17β-HSD), steroid 5α-reductase, 3α-hydroxysteroid dehydrogenase (3α-HSD), and aromatase [54]. The pilosebaceous unit and the sweat glands contribute to local synthesis of androgens and estrogens. Steroid sulfatase hydrolyses DHEAS to DHEA [55] (possibly in the sebocytes or in the dermal papillae of terminal hair follicles, which show enzymatic activity).
Within the pilosebaceous unit, testosterone is both produced from adrenal precursors as well as inactivated; this maintains androgen homeostasis [51, 52]. In sebocytes, the 1- isotype of 3β-HSD converts DHEA to androstenedione, and the 3- and 5- isotypes of 17β-HSD convert androstenedione to testosterone. Conversely, the 2- isotype (present in the root sheath cells of hair follicles) and the 4- isotype (in epidermal keratinocytes) deactivate testosterone in the reverse direction and play a protective role against androgen excess [54]. Keratinocytes also are responsible for androgen degradation [52].
Sebocytes are likely the major site of 5α-reductase activity in the skin. The type 1 isotype of 5α-reductase, expressed in sebaceous glands and sweat glands (with lesser activity in epidermal cells and hair follicles) [56], converts testosterone to the highly potent androgen, DHT. The type 2 isotype is active in beard hair follicles. A newly detected type 3 isotype, sensitive to finasteride (a 5α-reductase inhibitor), is strongly expressed in sebaceous gland cells [57]. Two isozymes of 3α-HSD deactivate DHT by conversion to 3α-androstanediol.
Aromatases in the sebaceous glands, the outer and inner root hair cells of anagen hair follicles, and dermal papillae cells [52, 53] convert androstenedione and testosterone to estrogens. Aromatase expression is much higher in scalp hair follicles of women than of men, and the enzyme is rarely expressed in telogen hair follicles [58].
11.3.4 Sex Steroid Receptor Localization in the Skin
Androgens (specifically testosterone and 5α-dihydrotestosterone (DHT)) and estrogens (specifically estradiol) mediate their skin effects by activating specific cellular receptors. Testosterone and DHT act through a single nuclear androgen receptor (AR), and their activity on the skin depends on receptor distribution. AR is present in epidermal and follicular keratinocytes, sebocytes, sweat glands, dermal papilla cells, dermal fibroblasts, endothelial cells, and genital melanocytes [59, 60].
Two distinct intracellular estrogen receptors, ERα and ERβ, belong to a superfamily of nuclear hormone receptors. Cell membrane-bound estrogen receptors also exist that activate signaling cascades via second messengers. ERβ is the predominant receptor in adult human scalp skin, strongly expressed in the stratum basale and stratum spinosum of the epidermis [59, 61]. ERα and ERβ are expressed in the sebaceous gland [59] and in primary cultures of dermal fibroblasts [62, 63].
Studies suggest that ERβ is the mediator of estrogen effects on skin and hair. ERβ is strongly expressed in anagen hair follicles of the human scalp, where it is localized to nuclei of the outer root sheath, epithelial matrix, and dermal papilla cells [59]. ERβ is also highly expressed in the epidermis, sebaceous glands, blood vessels, and dermal fibroblasts (see Table 11.3).
11.4 Effects of Sex Hormones on the Structure, Characteristics, and Physiology of Aging Skin
Clinically obvious signs of skin aging are wrinkling, pigmentary changes, loss of skin elasticity, and sagging. Skin exposed to UV damage has coarser and deeper wrinkles; a roughened, leathery surface; mottled pigmentation; and a more pronounced loss of elasticity. Intrinsically aged skin, which reflects purely chronological changes, has a dry, smoother texture, with fine wrinkles and an unblemished surface; loss of elasticity is less severe than in skin exposed to UV light. Aging skin is also thinner and more vulnerable to damage.
In addition to changes in structure, the growth rate of the hair and nails slows with age, the nail plate thins, and its surface becomes ridged and lusterless. The hair loses pigment and the density of hair follicles on the scalp decreases, independent of androgenetic alopecia (genetically driven patterns of balding). Vellus hairs in the ears, nose, and eyebrows of men and hair on the upper lip and chin of women convert to more obvious terminal hairs.
The influence of the sex steroids on these changes and the resulting gender differences in aging skin aging are summarized in Table 11.4 and reviewed below.
Table 11.4
Age-related changes in skin structure and physiology affected by the sex steroids in men and women
Parameter | Change | Gender differences | Impact of sex hormones |
|---|---|---|---|
Wrinkles | Develop and become more pronounced with age | No established gender differences | Wrinkling may be related to reduced stimulation of collagen and glycosaminoglycan synthesis by estrogen |
Skin thickness | Becomes thinner with age in both sexes (atrophy) | Skin of adult men is thicker than that of women [66] | Much of the decrease in skin thickness is thought to result from collagen changes in the dermis (see below) |
Epidermal thickness decreases 6.4 % per decade [67] | |||
Skin thickness decreases faster in older women than in older men [67] | |||
Dermal thickness decreases 20 % by old age [68] | |||
Collagen (dermis) | Fibers more disorganized; balance between synthesis and degradation shifts toward greater degradation [69] | See above | |
Elastin (dermis) | Fibers degrade; skin less elastic | Alterations more pronounced in older women [68] | |
Skin barrier function | No established gender differences [82] | ||
Skin moisture and water-holding capacity | Reduced water content of stratum corneum [88] | No established gender differences | |
Reduced water-holding capacity of the dermis due to declines in glycosaminoglycans and hyaluronic acid [93] | |||
Sweating and thermoregulation | Sweat glands express 5α-reductase (which converts androgens to DHT) and the androgen receptor through which DHT exerts its action | ||
Elevated temperature thresholds for sweating and reduced sweat response more pronounced in older women than in men [96] | |||
Sebum production | Gradual decrease in women | In men, sebum levels change minimally from puberty until about age 80 | Sebocytes regulate the effect of androgens in the skin |
Testosterone promotes DHT synthesis in sebocytes and stimulates sebum production [101] | |||
In older women, estrogen supplementation suppresses sebum production; progesterone overcomes this effect [102] | |||
In women, sebum secretion decreases gradually from menopause through age 80, after which no appreciable change occurs [104] | |||
Hair growth | Androgenetic alopecia usually begins around age 30 in genetically susceptible men and women | Male-pattern baldness is more common and severe and can start as early as late adolescence | Both androgens and estrogens affect hair growth. DHT acts on hair follicles to release growth factors in androgen-dependent areas (beard, axilla, pubis) [105] |
Female-pattern baldness is less common and usually milder | |||
In male androgenetic alopecia, DHT causes susceptible scalp follicles to miniaturize; the number of follicles in anagen phase decreases | |||
In women, scalp hair follicles have lower 5α-reductase levels, lower AR levels, and higher aromatase activity, limiting the impact of DHT | |||
Estrogens act on hair follicles through ERβ, which is present at sites of hair renewal in follicles of women but not of men | |||
Wound healing | Men display lower rates of wound healing at all ages | Androgens depress wound healing by increasing inflammation, proteolysis, and matrix degradation [108] | |
Estrogens promote wound healing by inhibiting inflammation and promoting keratinocyte mitogenesis, deposition of matrix components, and angiogenesis [109] | |||
Subcutaneous DHEA restores wound healing rates in ovariectomized mice and promoted wound healing in aged mice, likely through local conversion to estrogen [110] |
11.4.1 Skin Structure and Thickness
Human male skin is thicker and drier than female skin throughout the life span (from ages 5 to 90 years) [66, 111]. In part, this is because androgens stimulate epidermal hyperplasia in adult human skin [83]. Although the skin thins with age in both sexes, in men, skin thickness decreases linearly beginning at age 20, whereas in women, it remains relatively constant until about age 50 and then decreases [112]. Epidermal thickness decreases about 6.4 % per decade on average, but faster in postmenopausal women than in men [67]. Dermal thickness decreases by up to 20 % in both genders [68], although in sun-protected sites, significant dermal thinning occurs only after the eighth decade [113].
The decline in dermal thickness accounts for most of the measurable thinning of aging skin. The major extracellular components of the dermis (collagen, elastin, and hyaluronic acid) are affected by age. Collagen fibers become disorganized, especially in photoaged skin, as the matrix metalloproteinases, which degrade collagen, are upregulated by UV exposure [69]. When the balance between collagen synthesis and degradation is disturbed, the collagen fibers fragment, disrupting the tension on dermal fibroblasts that exists in a healthy collagen matrix and causing fibroblasts to collapse [74, 75].
Elastin calcifies and degrades with age and its turnover declines [114]. These changes make the skin less elastic, less extensible under force, and more vulnerable to injury by shear forces. These properties erode more dramatically in women than in men [68].
DHEA plays a role in maintaining skin structure. It regulates the synthesis and degradation of extracellular matrix protein; it promotes procollagen synthesis; and it limits collagen degradation by decreasing the synthesis of collagenase and matrix metalloproteases and increasing the production of tissue inhibitors of matrix metalloproteinase [72, 73]. Consequently, the substantial decline in DHEA with age reduces procollagen synthesis and elevates collagen degradation.
Oral DHEA treatment in men and women aged 60–79 for 1 year improved epidermal thickness and skin hydration, increased sebum production, and reduced facial pigmentation, with effects being more dramatic in women over 70 than in men [99]. DHEA is the primary source of sex steroid production in skin: because older women have lower circulating levels of DHEA than older men, they may have benefited to a relatively greater degree from DHEA supplementation.
Most studies on the effects of estrogen on aging skin have examined the uses of systemic or topical estrogens in postmenopausal women. Estrogen slows or reverses these manifestations of skin aging, maintaining skin thickness, collagen content, and hydration.
Estrogens affect skin thickness and elasticity primarily through their impact on constituents of the dermis. Hormone replacement therapy maintains or improves skin thickness following menopause, largely by affecting dermal thickness. For example, nuns treated with oral conjugated estrogens for 1 year experienced a significant increase in dermal and overall skin thickness compared to placebo-treated controls [115]. Other studies found that postmenopausal women receiving HRT achieved skin thickness levels comparable to those of premenopausal women [116].
Collagen reduction is a major factor responsible for skin atrophy. The declines in the quality of collagen and elastin with age are more pronounced in aging women, probably due to estrogen deficiency. After a slight delay following the onset of menopause [76], total collagen content declines an average of 2.1 %/year in the first 15 postmenopausal years [76, 117]. Clinical studies have demonstrated beneficial effects of oral, topical, and subcutaneous estrogen treatment on collagen content (reviewed in [118–120]). The benefits of HRT or estrogen supplementation on collagen content are proportional to baseline levels at the time of treatment [76, 117].
Estrogen also benefits skin elasticity. In the first 5 years following menopause, facial skin distensibility increases 1.1 %/year and elasticity decreases by 1.5 %/year [78, 79, 121]. Women who received HRT during this time period experienced no significant changes in skin elasticity. Studies of oral, transdermal, and topical estrogen treatment also showed benefits [79, 102, 122, 123], although topical estrogen treatment seems to be effective only in sun-protected skin [124]. The extent to which the effect is due to improvements in elastin fiber quality is unclear.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


