Introduction
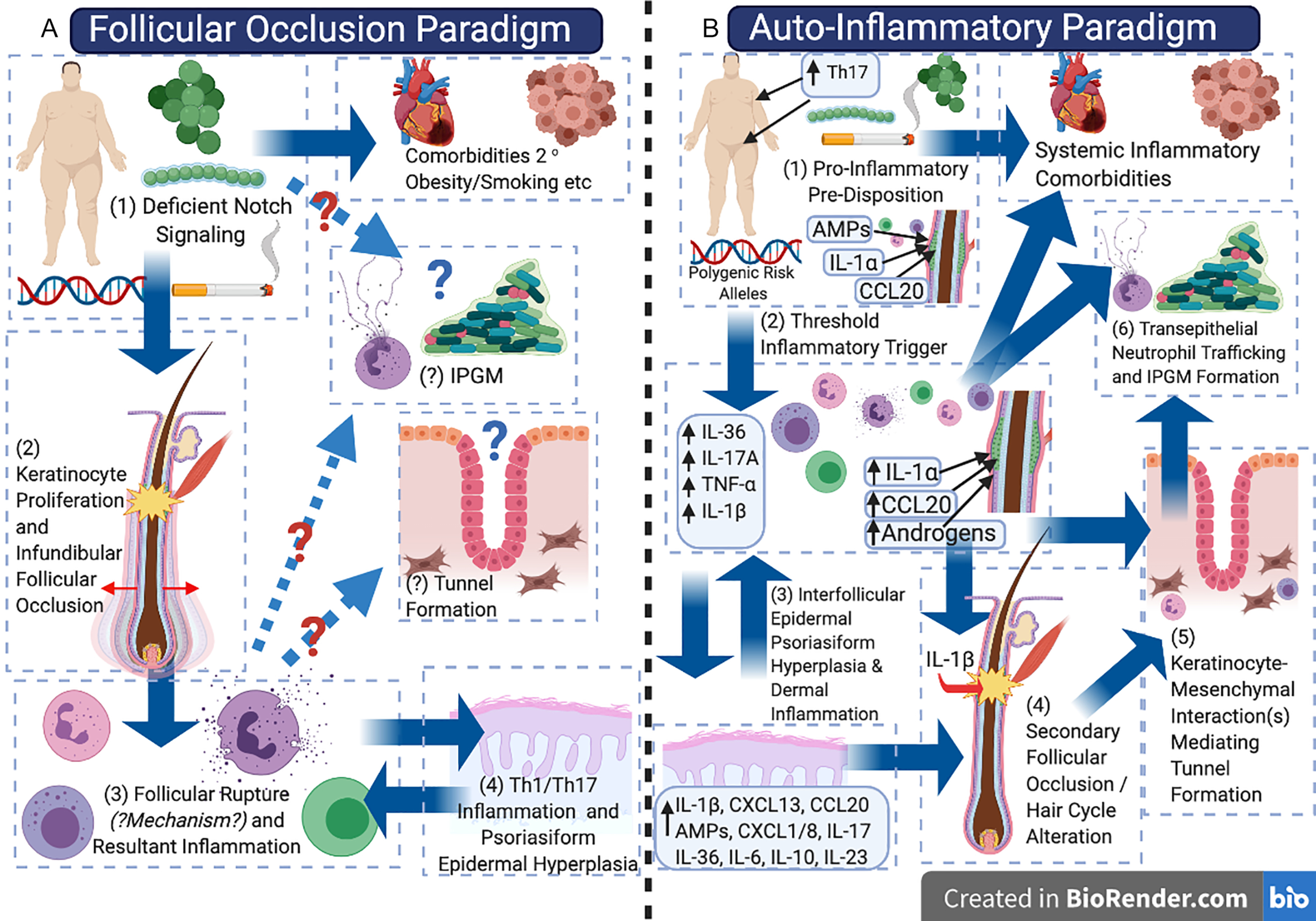
The pathogenic model of hidradenitis suppurativa (HS) may be in the midst of a paradigm shift, balancing the initial model of a disorder of (primary) follicular occlusion with consideration as an autoinflammatory keratinization disease (AiKD). There is observational, experimental, and therapeutic evidence to support the concept of HS as a primarily inflammatory disorder and/or a disorder of autoimmunity (in contrast to that primarily of follicular occlusion); however, the lack of reliable disease models has limited experimental/mechanistic evidence to support or refute one pathogenic model over another ( Fig. 10.1 ). Continual re-evaluation and integration of current clinical, histological, and molecular data into our pathogenic model of HS is essential in order to advance our understanding of the disease. Challenging existing paradigms through observation, hypothesis, and experimentation (while maintaining an open mind) is a core component of the scientific process, and is essential to enable accurate identification of novel therapeutic targets and treatment strategies. It is also vital to the understanding of differential treatment response in different individuals and exploring the potential role of variations in inflammatory endotypes (disease subtypes defined by a distinct functional or pathological mechanism) in the disease, in a similar way to how this has been identified in atopic dermatitis. This review aims to synthesize existing knowledge from clinical observation, classical histology, as well as modern molecular biology techniques to evaluate the evidence for HS as either a disorder of follicular occlusion or an AiKD.
The Evolving Pathogenic Paradigm(s) of Hidradenitis Suppurativa
Historically, HS has been proposed to be a disorder of apocrine gland inflammation, although multiple independent histological studies have demonstrated that inflammatory involvement of apocrine glands is a secondary phenomenon, and that the primary inflammatory driver of the disease exists adjacent to keratinocytes of the follicular infundibulum and interfollicular epidermis. It is now widely accepted that the primary driver of disease activity centers upon the follicular infundibulum. Additionally, other disorders such as pilonidal sinus disease and dissecting cellulitis of the scalp share many clinical, histological, and inflammatory features with HS and are beginning to be considered as uni-localized variants of disease. Melnik’s seminal 2013 paper began to shift the pathogenic paradigm of HS away from an apocrine-gland based inflammatory or infectious disorder, to a disorder of follicular occlusion and proposed dysregulated Notch signaling as the unifying feature of HS pathogenesis.
Emerging evidence as to the role of the inflammasome, complement, and IL-1 isoforms has led to the suggestion of HS as an AiKD. Evidence of systemic inflammation, activation of B cells, and plasma cells have raised the possibility of HS having an autoimmune or antibody-mediated component. However, follicular occlusion is still considered the “primem movens” of HS preceding the inflammatory drive of disease.
The Problem(s) with Animal and Ex-Vivo Models in Hidradenitis Suppurativa
The development of animal models in HS dates from the identification of gamma secretase associated polymorphisms in familial HS. Gamma secretase and Notch-1 null mice were incompatible with life due to the vital role of notch signaling in body segment development; however, post-natal knockdown of Notch-1 results in hyperproliferative epidermis, hair loss, and epidermal cyst formation in adult mice. Combined Notch-1 and Notch-2 knockdown results in an atopic-dermatitis-like epidermal disorder and a rapidly fatal myeloproliferative disorder in adult mice. When interpreting these results through the follicular occlusion paradigm, it is reasonable to assume that epidermal cyst formation is consistent with the proposed initial steps in HS pathogenesis; however, the lack of inflammation, cyst rupture or tunnel development argue against Notch-1 knockdown being a high fidelity animal model for HS.
One possible explanation for the lack of comparable dermal inflammation and tunnel formation in Notch-1 knockdown murine models may be the difference in dermal thickness between human and murine skin and the differential localization of the follicular unit in the dermis (human) and subcutis (murine). In order to overcome these potential issues, a murine xenotransplantation model was developed involving direct transplantation of lesional HS tissue to immunosuppressed mice. The most recent model developed involves a transwell ex-vivo model, which overcomes the structural and morphological issues of dermal thickness and follicular insertion of murine models and has relatively less infection risk than xenotransplantation models. However, given the overlapping limitations of existing models, only translational studies of interventions in human subjects give the greatest fidelity in both mechanistic and clinically relevant responses to pharmacological interventions in HS in order to explore the pathogenic mechanisms of disease.
Follicular Occlusion: Comedones are Clinically and Experimentally a Product of Inflammation, Rather Than a Cause
Histological studies illustrate the prominent roles of comedogenesis, follicular hyperkeratosis, and comedogenesis in HS tissues. However, in each instance, the coexistence of perifollicular inflammation is comparably prominent. Clinically, comedones (both open and closed), as well as typical double-sided comedones, are present in diseased areas, inflamed tissues, and also in scarred, non-inflamed tissues. They are also present in areas not exposed to flexural occlusion.
Von Laffert et al. report comedones as more common in end stage fibrotic and scarred lesions and independent of the follicular unit. These comedones are more likely to be those of the double-ended variety which were once considered to be pathognomonic of HS. From these clinical observations, it can be concluded that comedones are associated with HS; however, the establishment of causation requires mechanistic evidence. Such mechanistic evidence is available thanks to investigations into comedogenesis in acne research. Recent findings have identified subclinical inflammation as preceding comedogenesis in acne prone skin, disrupting the longstanding assumption that follicular occlusion is the primary initiating factor in acne. The molecular mechanisms of comedogenesis involve follicular keratinocytes, producing a number of pro-inflammatory mediators (including antimicrobial peptides, microbial associated proteins including lipotechoic acid, CCL20, and IL-1α). Ex-vivo studies of the follicular infundibum isolated in vitro are able to recapitulate the formation of comedones with addition of IL-1α and prevent formation with the addition of IL-1RA. It is acknowledged that the in vitro studies performed are based on highly sebaceous follicular units which have distinct differences from apocrine bearing skin ; however, the similarities in immunological milieu between sebaceous and apocrine skin in Th17 associated mediators (central to inflammation in HS) raise the theoretical possibility of these mechanisms being shared between body sites. Reproduction of these experiments using follicular infundibular from apocrine gland bearing regions would hopefully be definitive in confirming or refuting these findings.
Therefore, it can be concluded that molecular and ex-vivo evidence suggests comedone formation is possible secondary to subclinical inflammation, rather than inflammation being the result of comedone formation and follicular rupture. The precise mechanisms that have been demonstrated in human skin, however, require validation in apocrine gland bearing skin given the unique immunological milieu of these sites. These results may explain the diffuse scattering of comedones seen in HS prone areas, the presence of comedones in extra-flexural sites, and the presence of comedones in previously inflamed (“burnt out”) tissue or sites distant from a follicular unit. It also raises the additional question of what differentiates conditions in which subclinical inflammation and diffuse flexural comedone formation (such as Dowling-Degos disease ) exist, from highly inflammatory HS lesions. Direct molecular comparisons of these conditions may further inform the differences in subclinical inflammation that leads to the development of one condition over another (or the coexistence of both conditions at different timepoints).
Skin Fold Occlusion is Associated with Microbiome Alterations and Subsequent Pro-Inflammatory Keratinocyte Responses
On a microscopic level, follicular occlusion in HS refers to occlusion of the follicle at the infundibulum. However, from a clinical viewpoint, “follicular occlusion” may refer to the anatomical sites of predilection in HS: namely axillary, inguinal, and sub-mammary folds. These areas are subjected to friction, skin-to-skin contact, and well-documented alterations in moisture, pH, and microbiological colonization ( Fig. 10.2 A and B ). In the setting of obesity, the posterior neck folds, abdominal pannus, gluteal cleft and inner thighs, and other anatomical sites can undergo similar microbiological milieu alterations secondary to heat, moisture, and pH changes.
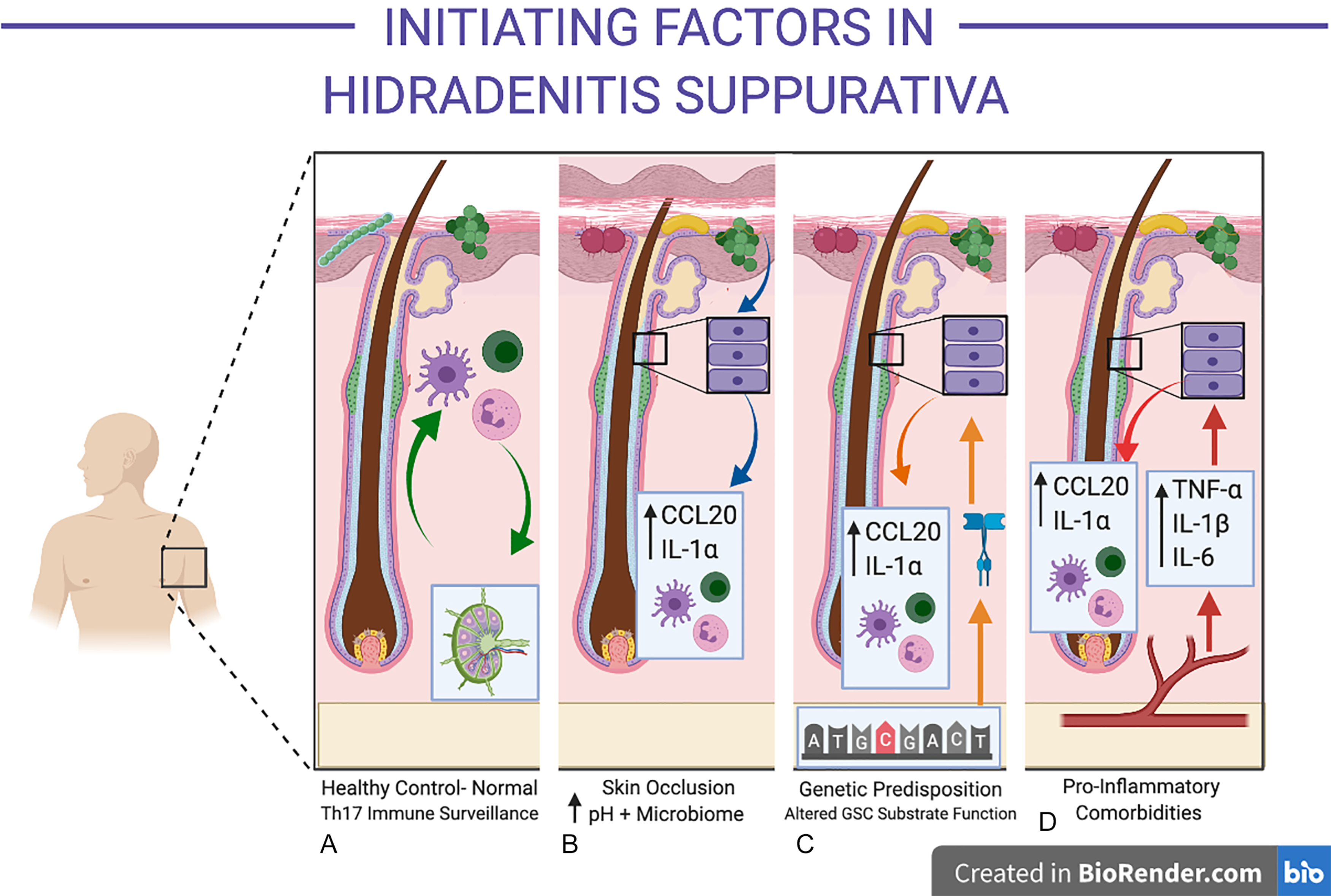
The follicular infundibulum is an immunologically active, microbially colonized site, involved in the development of immune tolerance to commensal organisms. This differs substantially from other portions of the follicle (such as the bulb), which are considered immunologically privileged sites. Infundibular keratinocytes produce CCL20 and antimicrobial peptides under normal physiological conditions. Increasing moisture decreases the pH of the stratum corneum and pH reductions are known to promote the colonization and activity of porphyromonas sp (a well-documented microbiont-associated with HS). Other bacterial species including Staphylococcus aureus Propionibacterium acnes, yeasts, and their associated proteins (including lipoteichoic acid) are able to induce the release of pre-formed IL-1α in keratinocytes. Indirect evidence for the role of yeasts in inflammatory activity in HS is demonstrated in recent observational studies of anti- Saccharomyces cerevisae antibodies in severe HS, which can also cross-react to other fungal and bacterial species. While the precise mechanisms of specific microbiological species and/or strains in HS are ill-defined, their functional role in producing an aberrant pro-inflammatory response (either directly or indirectly via keratinocytes) is consistent with observational studies identifying these microbionts in both early and advanced disease.
Genetic Variants in HS May Act via EGFR-Associated Pathways Linking Follicles, Th17 Mediated Inflammation, and Drug-Induced Disease
Chapter 12 summarizes the published literature on genetics and epigenetics of HS. The role of inherited mutations in Notch signaling as the pathogenic mechanism in HS has come under scrutiny, with the existing genetic and molecular evidence suggesting a more complex interplay between genetics and infundibular keratinocyte-derived inflammation (see Fig. 10.2 C).
The first documented mutation in familial HS was in Nicastrin, a component of the gamma secretase complex (GSC), identified in 2010 in an East-Asian kindred. Mutations in the GSC are also associated with familial Alzheimer’s disease and cardiomyopathy, although no common variants with HS are known. Since then, a small minority of patients with familial and spontaneous HS have been identified with GSC mutations. This suggests that other identified loci may contribute to genetic predisposition to HS, although this will only be elucidated with the results of genome wide association studies in HS. The precise mechanism of action of GSC mutations in the pathogenesis of HS is unclear. The GSC complex cleaves over 70 different substrates involved in cell cycle and inflammation including EGFR, IL-1, TNF-α, complement regulatory protein CD46, and Notch. Melnik’s seminal 2013 paper proposed Notch signaling as the unifying motif in HS pathogenesis via associations with keratinocyte proliferation, smoking, and sequence variants in GSC. The molecular evidence for Notch being associated with keratinocyte hyperproliferation is well established ; however, dysregulated Notch signaling is also associated with other inflammatory skin disorders including psoriasis, atopic dermatitis, and alopecia areata. Notch dysregulation may be an epiphenomenon secondary to keratinocyte proliferation (as it is present in multiple other inflammatory dermatoses) rather than the primary cause of HS.
In-silico evidence has identified HS-specific GSC substrates ERbb4 and Tie1 as differentially expressed substrates that distinguish the transcriptome of HS from familial Alzheimer’s disease and other inflammatory skin diseases. ErbB4 and Tie1 are components of the EGFR pathway (active in the follicular infundibulum) and are associated with SOX9 and Wnt signaling linked with hair cycle progression, IL-17A production (through shared downstream Act1 activity), and epithelial cell fate, all mechanisms identified in transcriptomic analysis of HS tissues.
In vitro studies have demonstrated diverse pro-inflammatory results of Nicastrin knockdown including IL-36a production, alterations in EGFR signaling, as well as increased sensitivity to interferon mediated pro-inflammatory pathways. Recently, mutations in POFUT1 have been identified in cases of Dowling-Degos Disease associated with HS. POFUT-1 is a fucosyltransferase which is active upon multiple substrates including Notch and EGFR and is important for post-translational modification of receptors. Therefore, abnormal activity of the EGFR pathway is linked with infundibular keratinocyte differentiation and Th17 inflammatory pathways. The link to clinical disease activity is supported by reports of HS associated with use of EGFR antagonists in oncology. Therefore, dysregulation of EGFR associated pathways secondary to GSC mutations may explain both the infundibular localization of HS, the involvement of the Th17 immune axis, and cases of HS-like features in the setting of EGFR antagonism.
Disease Initiation is Associated with Systemic Subclinical Inflammation and Dysregulated Infundibular Keratinocytes
The site of initial inflammation in HS is centered upon the infundibulum of the hair follicle. Given the active immunologic role of the follicular infundibulum, a degree of baseline inflammatory activity around the follicle is considered normal. Understanding of the initiating factors associated with the excessive and self-perpetuating peri-follicular inflammation in HS remains incomplete (see Fig. 10.2 D). Epidemiological and clinical observations suggest that a number of systemic disorders (including insulin resistance, hormonal dysregulation, and obesity) may be associated with HS and contribute to a pro-inflammatory state. In other inflammatory disorders such as psoriasis, rheumatoid arthritis and atherosclerosis, these factors have been found to be associated. However, the causation between disease and systemic inflammation is still a topic of contention and is in need of further mechanistic enquiry.
Regardless of the direction of causation between HS comorbidities and inflammation, guidelines and limited clinical evidence suggest benefits to weight loss, smoking cessation, and dietary counseling as an integral part of HS management. The mechanisms of these pro-inflammatory cascades are complex and incompletely understood. Smoking, via polycyclic aromatic hydrocarbons, can directly alter follicular keratinocyte differentiation resulting in comedogenesis. It can also produce widespread methylation changes and systemic increases in IL-6, C-Reactive Protein (CRP), fibrinogen, and multiple members of the NF-κB family. Adipose tissue can produce pro-inflammatory signatures including IL-6, IL-1β, and TNF-α in the setting of chronic nutrient excess. Additionally, adipokines can mediate both inflammation and the development of insulin resistance, which is also associated with HS. Keratinocytes in the infra-infundibulum of the follicle express Type 1 5-hydroxy-testosterone ; modulating infundibular keratinocyte differentiation programs both directly as well as via fibroblast activation and fibroblast-keratinocyte interactions, contributing to androgen-induced follicular changes.
Overall, these associations suggest that a systemic pro-inflammatory state and localized infundibular keratinocyte dysregulation are potential predisposing factors to clinical disease. There are contradictory reports pertaining to the benefit of withdrawing these predisposing factors (e.g., cessation of smoking/weight loss) during established disease. These findings only appear contradictory if one holds the assumption that the initiating and perpetuating factors of clinical disease in HS are one and the same. As other authors have suggested, there may be unique factors contributing to each state (initiation of disease and perpetuation of disease), and our lack of data regarding early (subclinical) disease has not allowed us to appreciate this fact.
Inflammation in Hidradenitis Suppurativa: Evidence from Existing Studies
The inflammatory signature of established HS has been well characterized in multiple histological and molecular studies. Similarities and parallels with psoriasis have been observed in lesional and perilesional HS tissue, with lesional nodules demonstrating mixed inflammatory infiltrates comprising of T cell, dendritic cells, plasma cells, neutrophils, and monocytes. Chronic longstanding disease also demonstrates B cell infiltrates, NETosis, and development of epithelialized tunnels. An issue with understanding the characteristics of inflammation in HS is that the majority of specimens isolated for studies are from those with severe, longstanding disease. Hence, we have limited insight into the initiating events in early and/or mild HS. Additionally, until recently, there were no standardized, defined biopsy sites for investigational studies. Given that HS is morphologically diverse, it would be erroneous to assume that a biopsy from one portion of tissue is representative of all the different epidermal (and deep dermal) morphologies present across the spectrum of HS. Therefore, studies which do not define the severity, treatments, sites, and lesion types of biopsies should be interpreted with caution.
The common inflammatory signatures identified by qRT-PCR studies in HS lesional tissue include TNF-α, IL-1α, IL-1β, IL-6, IL-17A, IL-17F, IL-32, IL-36α, IL-36g, and IL-10. Additional chemokines include CCL3, CCL5, CCL27, and BLC. Non-lesional tissue also demonstrates upregulated levels of many of these cytokines, although variation does exist due to previous lack of standardized biopsy sites and combination of both partially treated as well as untreated specimens. Transcriptomic studies demonstrate strong B-cell signatures with IgG1 and IgG3 immunoglobulins and aspects of the complement cascade highly upregulated. Additionally, signals of keratinocyte hyperplasia (Keratin 6, Keratin 16) are also seen with keratinocyte derived factors being elevated in lesional and perilesional tissue compared with unaffected and control skin. Variation in cytokine levels do occur (between lesional, perilesional, and non-lesional tissue) in terms of type and degree of inflammation, although reliable characterization of inflammation matched with disease morphology (e.g., nodules vs. tunnels) is yet to be undertaken. Scarred tissue demonstrates decreased inflammatory profiles compared to non-scared areas, and the presence of occult dermal tunnels can also induce highly inflammatory profiles in normal-appearing skin. The use of clinical ultrasound has been suggested as a method of confirming or excluding the presence of tunnels prior to biopsy.
Analysis of serum has identified IL-1β, IL-6, IL-8, IL-10, IL-12p70, IL-17, and TNF-α as upregulated in multiple studies; however, conflicting results exist between serum levels of IL-10, IL-17, and IFN-y, which may be secondary to the severity of included participants and the methods of cytokine analysis. The majority of data regarding serum inflammation is based on patients with Hurley stage 2 and 3 disease, with the changes in serum inflammatory markers in early and mild disease unclear.
In terms of establishing mechanism—it has been assumed, based on observational studies, that perilesional inflammation is of the same character (albeit less intense) as nearby lesional inflammation. Therefore, given the known feedback mechanisms between IL-1 and IL-17 leading to self-perpetuating feed-forward inflammation, it is reasonable to assume that lesional tissue inflammatory characteristics may be replicated by adding pro-inflammatory cytokines to ex-vivo perilesional tissue. This experiment (conducted by Vossen et al. ) was unable to replicate the lesional HS inflammatory profile, suggesting that IL-1α and/or IL-1β are not the sole triggers necessary to induce development of lesions in HS. Other possibilities are that a combination of multiple inflammatory mediators are required; or as-yet-unknown predisposing factors are involved in inducing active inflammatory nodules on a background of perilesional subclinical inflammation. This raises the prospect that the process of inflammation in HS is more complex than initially thought. The underlying assumption thus far in HS research is that perilesional tissue represents the same inflammatory profile as lesional tissue, differing only in the degree, intensity, and more superficial location of inflammation. However, an alternative hypothesis that the inflammatory characteristics of perilesional tissue are distinct from lesional tissue remains to be thoroughly investigated.
TH17 Feed-Forward Inflammation is Prominent in Established Disease
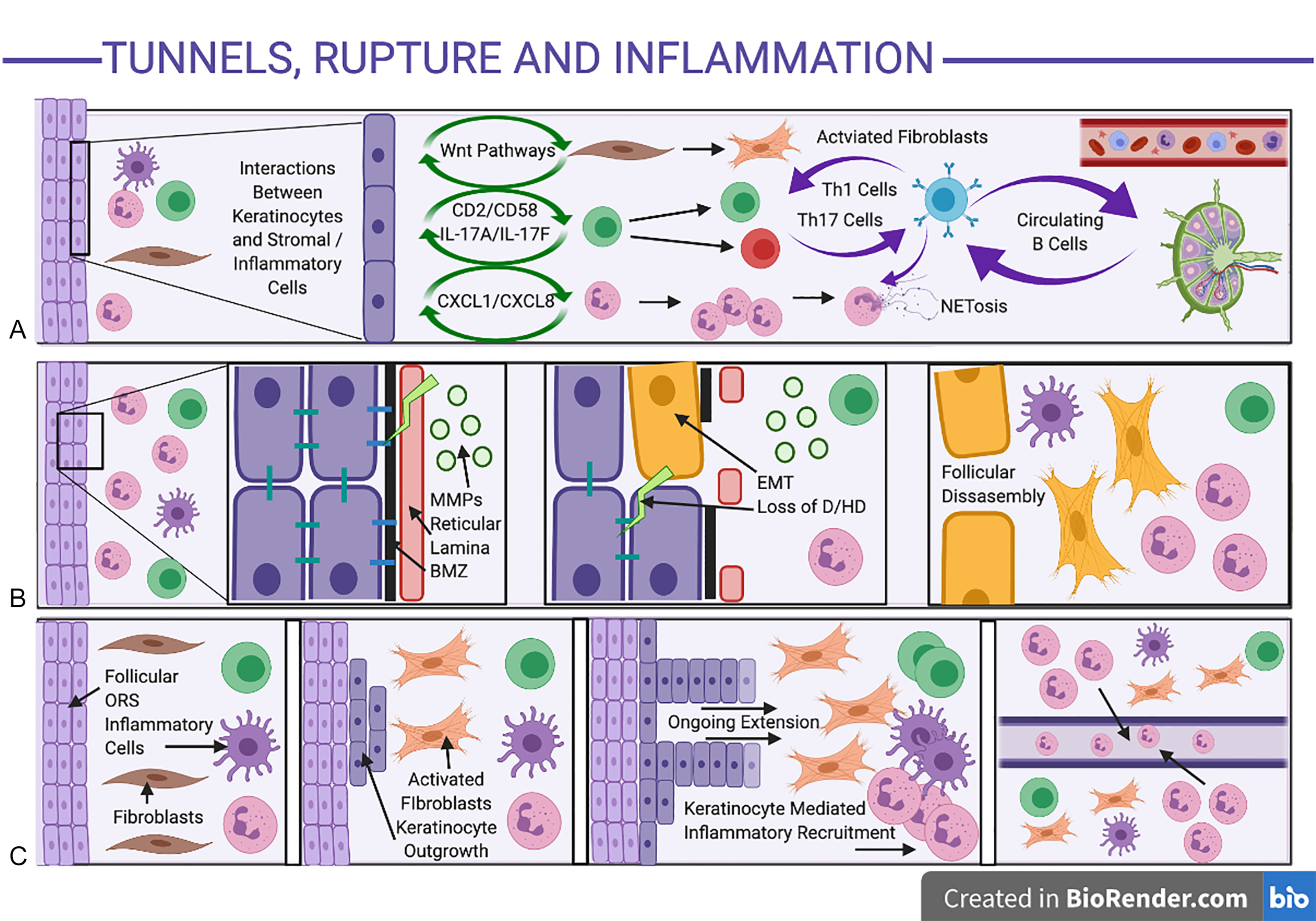
The Th17 axis is strongly implicated in established self-perpetuating clinical disease ; however, the mechanisms leading to Th17 feed-forward self-amplification in HS are still unclear. It is assumed to be similar to the activation of the Th17 axis in psoriasis, with the predisposition of the axillae and other areas of apocrine gland-rich skin to a Th17 immune response as demonstrated experimentally. There is well-documented evidence (largely from the psoriasis literature) regarding positive feedback loops (“feed-forward mechanisms”) between IL-1β, IL-6, and TNF-α by IL-17, leading to further IL-1β, IL-6, and TNF-α production as well as downstream activation of acute phase reactants, neutrophilic, and complement mediated inflammatory responses. This is perpetuated through leucocyte-keratinocyte interactions, amplifying antimicrobial peptide and chemokine production (including CXCL1 and CXCL8), leading to additional inflammatory cell recruitment adjacent to IL-17-activated epidermal keratinocytes ( Fig. 10.3 A ). Such inflammatory cell localization has been seen surrounding intrafollicular and interfollicular sites adjacent to epidermal keratinocytes in early histological specimens of HS, with evidence of early psoriasiform hyperplasia suggestive of IL-17-induced epidermal changes. Despite the majority of translational work focusing upon IL-17A (given the body of pre-existing work based in psoriasis), significant elevations of other IL-17 isoforms including IL-17C and IL-17F are seen in HS tissue 32,108 and may be significant contributors to disease activity which are not targeted by anti-IL-17A therapies alone.