Chapter 31 Etiology and prevention of multisystem organ failure
 Access the complete reference list online at http://www.expertconsult.com
Access the complete reference list online at http://www.expertconsult.com
 IN THIS CHAPTER
IN THIS CHAPTER  PowerPoint Presentation Online
PowerPoint Presentation Online
Introduction
Despite its common occurrence in critically ill burn patients, our understanding of the multisystem organ failure syndrome remains fragmented and incomplete. The cascade of organ dysfunctions which typify the multisystem organ failure syndrome is driven by an unregulated inflammatory state, often, but not always, associated with uncontrolled infection.1 Other potential ‘engines’ driving this cascade of organ dysfunctions are an impaired gastrointestinal barrier,2,3 the open burn wound,4 and inadequate delivery of oxygen to peripheral tissues.5 The line between organ dysfunction and failure is admittedly unclear, but a set of organ-specific definitions of failure is helpful and has been developed (Box 31.1).6 Approximately 15% of patients admitted to surgical intensive care units have multisystem organ failure,7 and perhaps 8% of burn patients ultimately develop the syndrome.8
Box 31.1 Organ-specific definitions of failure (OSF)
Neurological failure
• Glasgow Coma Score ≤6 (in absence of sedation at any one point in day)
• Glasgow Coma Score: sum of best eye opening, best verbal, and best motor responses.
Scoring of responses are as follows: (points)
The sequence of failures often follows a predictable course, although the cascade can be modified by various treatments, such as the prophylactic use of proton-pump inhibitors or H2-receptor blockers. In burn patients, two cascades have been described.8 An early clinical sequence is characterized by resuscitation failure, adult respiratory distress syndrome, hemodynamic failure, renal failure, liver failure, gut failure, and infection, and a later cascade typified by pulmonary failure, hemodynamic instability, renal failure, gut failure, and liver failure. Frequently, vasomotor failure and death is seen at the end of both cascades. Mortality increases with increasing numbers of failed organ systems. When three organs have failed, mortality has been reported as 100%, although survival is sometimes seen in patients with even more failed organ systems.9 An understanding of the progression of the syndrome aids in prognostication and facilitates decisions regarding termination of futile efforts.10,11 The management of specific organ failures will be presented in the next chapter. The purpose of this chapter is to discuss the etiology and prevention of the syndrome.
Etiology
Sepsis is clearly the most common initiator of the syndrome, and was recognized early on as the primary cause.1 One single overwhelming infection is not required, as small repetitive infections may initiate the cascade,12 perhaps by priming immune cells, making them react more intensely to each subsequent stimulus.12 It was recognized later that many patients with multisystem organ failure did not have infection,13 and this led the search for other ‘engines’. Endotoxin liberated from the walls of Gram-negative bacteria is a major, but not the only, intermediary,14 as Gram-positive bacteria cause similar aberrations in oxygen transport and hemodynamics,15 via similar cascades of mediators.16
In burn patients the wound may also be a source of the inflammatory mediators leading to multiple organ failure. Certainly, an infected wound will do this, but wound sepsis is decreasing in incidence with the advent of early burn wound excision17 and most infectious deaths in burn patients today are caused by pneumonia rather than wound sepsis.18 Complete wound closure, without donor sites, decreases oxygen consumption19 and presumably ameliorates the inflammatory response to the open wound. Incomplete wound closure does not have this effect.20 Increased levels of circulating mediators such as interleukin-6 (IL-6), IL-8, and tumor necrosis factor (TNF) have been shown to originate from the burn wound21 and contribute to the hypermetabolic and inflammatory state seen in burn patients. Interleukin-8 has been demonstrated to be upregulated in the lung after burn injury21 and the stimulus for this upregulation, which is associated with pulmonary dysfunction, may come from the wound.21
Failure of the gut barrier can contribute to the development of multisystem organ failure.22 Normal barrier function prevents the movement of bacteria and their products from the gut lumen into the portal and lymphatic circulations. Bacterial densities range from near 0 in the stomach, to 104–105 in the distal small bowel, to 1011–1012/g of stool in the normal colon.23 That the normal gut can carry this bacterial load, without the frequent occurrence of Gram-negative infection, is a tribute to normal barrier function. Although not seen immediately after trauma,24 several insults have been shown to result in increased translocation of bacteria and their products into the portal and lymphatic circulations. Hemorrhagic shock,25 endotoxin administration,26 burns,27 and burn wound sepsis28 have each been shown to result in increased translocation of bacteria from the gut. Gut permeability to macromolecules, such as endotoxin, has been shown to increase with increasing burn wound size using polyethylene glycol 3350 as a tracer.29 Smaller molecules, with lactulose as the tracer, have also been shown to pass more readily through the gastrointestinal membrane after injury.30 The exact mechanism by which bacteria and their products pass through the gastrointestinal barrier is not clear. Both intra- and transcellular processes may be involved.31,32 The consequences of loss of the gastrointestinal barrier are profound. Translocating whole bacteria can be a direct source of sepsis or can activate Kupffer cells2,3 and promulgate an inflammatory response in conjunction with bacterial products such as endotoxin.
Cellular dysfunction, caused by inadequate oxidative metabolism secondary to hypoperfusion, is another potential ‘engine’ resulting in multisystem organ failure. In ischemia-reperfusion models, oxygen radicals are generated, resulting in peroxidation of cell membrane lipids and accumulation of activated neutrophils,33 with progressive cellular and whole-organ dysfunction. It has been proposed that critically ill patients suffer from supply-dependent oxygen consumption because of defects in cellular oxygen extraction and utilization.34,35 This results in inadequate aerobic metabolism unless supranormal levels of oxygen are supplied.36 The reality of this proposal is still actively debated.37,38 Certainly, a grossly inadequate amount of oxygen available to cells dependent on aerobic metabolism can lead to cellular dysfunction, and this may be followed by organ failures.5 Maintaining oxygen delivery at least at normal, and possibly supranormal, levels should help maintain cellular homeostasis and minimize the risk of multiple organ failure.39 Although the requirement for a supranormal level of tissue oxygenation in critical illness is controversial and our ability to predict organ-specific oxygen delivery and consumption from whole-body data is poor,40 careful attention to whole-body hemodynamics is an integral part of the management of any critically ill patient.
Common ground: mediators
Sepsis, open burn wounds, impaired gut barrier function, and hypoperfusion are all associated with multiorgan failure. The similarity in the response to these differing events implies that there is some common ground. These processes all probably impact on individual organs via a number of mediators, whose complex interactions are still very poorly understood,41 but are being unraveled by investigators using blocking antibodies, soluble receptors, and receptor antagonists.42 At this point, these mediators, including endotoxin, arachidonic acid metabolites, cytokines, platelet activating factor, activated neutrophils and adherence molecules, nitric oxide, complement, and oxygen free radicals will be reviewed briefly.
Endotoxin, a lipopolysaccharide component of Gram-negative bacterial cell walls, induces many of the symptoms associated with sepsis, including fever, hypotension, the release of acute phase proteins, and the production of multiple cytokines including TNF and IL-1.43 Endotoxin also activates complement,44 causes activation of the coagulation cascade,45 and results in the release of platelet activating factor.46 Potential sources of endotoxin include both Gram-negative bacteria in foci of infection and Gram-negative bacteria within the gut when the gut barrier fails.
Arachidonic acid makes up approximately 20% of cell membranes, is released from these membranes in response to a multitude of stimuli which activate phospholipases A2 and C, and is then metabolized by one of two major enzyme systems (Fig. 31.1).
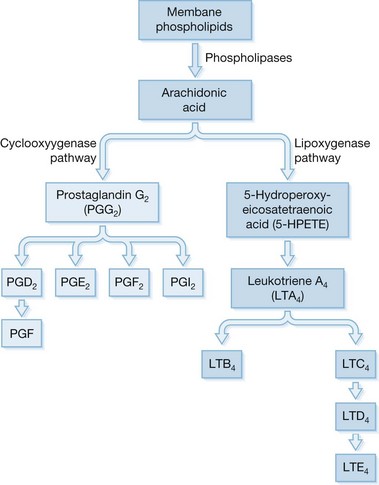
The cyclooxygenase pathway results in production of prostaglandins and thromboxanes, and the lipoxygenase pathway results in the production of leukotrienes.47 The prostaglandins and leukotrienes interact with other mediators in a complex fashion, and are later degraded by enzyme systems, which are dispersed throughout the body.48
Arachidonic acid, which is metabolized via the cyclooxygenase pathway, results in the formation of prostaglandins and thromboxanes. Prostacyclin inhibits platelet aggregation, thrombis formation, and gastric secretion.49 Thromboxane A2 (TXA2) causes platelet aggregation, has profound vasoconstricting effects on both the splanchnic and pulmonary microvasculature, and causes bronchoconstriction and increased membrane permeability.50 It is studied via its longer-lived but inactive metabolite, TXB2. Aspirin has its physiological effects by inhibiting thromboxane synthetase.51
Arachidonic acid metabolized via the lipoxygenase pathway results in the formation of leukotrienes. There are two types based on their metabolism after the action of 5-lipoxygenase, leukotrienes (LT) C4, D4, and E4 (the sulfidopeptide group) and LTB4.52 Leukotrienes are generated in response to multiple stimuli by several cell types, including neutrophils, macrophages, and monocytes.53 Vessel walls are also capable of generating leukotrienes.54 Leukotrienes C4, D4, and E4 have variable actions on vascular tone depending on the presence or absence of other mediators such as cyclooxygenase product.55 In addition to their variable effects in redirecting blood flow, LTC4, LTD4, and LTE4 also increase vascular permeability,56 and have been described as being elevated immediately prior to the development of pulmonary failure.57 The major effect of LTB4 is an enhancement of neutrophil chemotaxis.58 Thus, leukotrienes as a group may be involved in the edema formation and pulmonary and systemic vascular changes that are seen in the multisystem organ failure syndrome.
Cytokines are regulatory proteins that are secreted by immune cells and have multiple paracrine and endocrine effects. There are six major classes,59 including interleukins, TNF, interferons, colony-stimulating factors, chemotactic factors, and growth factors. Those which have been most extensively characterized are IL-1, IL-6, and TNF.
Interleukin-1 and IL-6 are elevated in septic states, and high levels are associated with a fatal outcome60 and predict systemic infection.61 Interleukin-1β causes hypotension and decreased systemic vascular resistance, which may be synergistic with the effects of TNF.62 Even better characterized is TNF, the administration of which causes hypotension, cardiac depression, and pulmonary dysfunction in animals.63–65 When administered to humans, TNF causes fever, hypotension, decreased systemic vascular resistance, increased protein turnover, elevation of stress hormone levels,66,67 and activation of the coagulation cascade.68
Platelet activating factor is a nonprotein phospholipid that is secreted by many cells including platelets, endothelial cells, and inflammatory cells,69 and is a major mediator of the pulmonary70 and hemodynamic71 effects of endotoxin. The major effects of platelet activating factor are vasodilation, cardiac depression, and enhancement of capillary leak. Its complex interactions with other mediators are still poorly understood.
Although tissue injury can occur in the absence of neutrophils,72 the inflammatory process results in local accumulation of activated inflammatory cells which release various local toxins such as oxygen radicals, proteases, eicosanoids, platelet activating factor, and other substances. When unregulated, such accumulations of activated cells can cause tissue injury.73 The initial attachment of neutrophils to the vascular endothelium at an inflammatory site is facilitated by the interaction of adherence molecules on the neutrophil and endothelial cell surfaces.74
These neutrophil adherence receptors are induced by numerous stimuli but, interestingly, are reduced after major thermal and nonthermal injury,75 perhaps explaining in part the increased incidence of infection in such patients. The importance of this adherence mechanism can be seen in patients who are deficient in one of the integrin class of neutrophil adherence receptors, CD-18, who suffer from frequent bacterial infections.76 The biology of the transmembrane polypeptides that govern these complex cell-to-cell interactions is an active area of research77 and holds promise for therapeutic interventions in the future.
Oxygen radicals, such as hydrogen peroxide and superoxide anion, are released by activated neutrophils in response to a variety of stimuli.78 They are also released when xanthine oxidase is activated after reperfusion in ischemia-reperfusion models. These highly reactive products cause cell membrane dysfunction, increased vascular permeability, and release of eicosanoids.
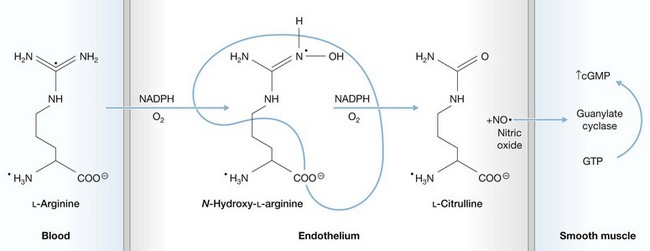
Nitric oxide, released when citrulline is formed from arginine (Fig. 31.2), was only identified as an endothelial product in the middle 1980s.79 Its half-life is only a few seconds, as it is quickly oxidized, but it has profound local microvascular effects. Nitric oxide synthesis is stimulated by various cytokines, endotoxin, thrombin, and by injury to the vascular endothelium. It is a potent vasodilator,80 but its actions vary depending on the vascular bed and presence of other mediators.81 Nitric oxide is one of the major mediators of the hypotensive response to sepsis.82,83
Antigen–antibody complexes activate the complement cascade, and complement fragments thus generated can interact with other cytokines to promulgate the inflammatory response.84 Diminished levels of a natural inhibitor of C5a have been demonstrated in patients with adult respiratory distress syndrome (ARDS)85 and administration of anti-C5a antibody diminishes hypotension in an animal model of endotoxemia.86 Complement fragments may be involved in the development of burn wound edema.87
Prevention of multisystem organ failure
Once established, multisystem organ failure is very difficult to reverse, emphasizing the importance of prevention.88 Prevention is based on our crude knowledge of the ‘engines’ that drive the process – sepsis, the gut, the wound, and inadequate perfusion (Table 31.1). Dealing with these issues is far more practical than dealing with an incompletely understood complex web of mediators. The discussion that follows will address prevention of sepsis via proper wound management and attention to unusual causes of sepsis, support of the gut, and prevention of inadequate oxygen delivery. Subsequently, the potential role of nutritional and specific immunomodulators will be addressed.
Table 31.1 Multiple organ failure etiology and established preventive measures
| Etiology | Prevention |
|---|---|
| Sepsis | Early excision and biologic closure of deep wounds |
| Anticipation and early treatment of occult septic foci | |
| Gut barrier failure | Optimize whole-body hemodynamics |
| Early enteral feedings | |
| Reduced organ perfusion | Optimize whole-body hemodynamics |
| Enhanced oxygen delivery |
Prevention of sepsis
In the burn patient, prevention of multisystem organ failure is greatly facilitated by prevention of wound sepsis via an aggressive surgical approach to deep wounds. The increasing survival of burn patients has paralleled the evolution of this approach to the burn wound.89–91 Not only is overt wound sepsis prevented by early removal of devitalized tissue, but multiple smaller septic insults are also prevented, as manipulation of heavily colonized burn wounds is a frequent source of transient bacteremia.92,93 Such multiple occult bacteremias occurring during frequent manipulation of heavily colonized wounds could contribute to the development of multisystem organ failure by priming immune cells, making them react more intensively to each subsequent insult.12 The role of perioperative antibiotics in minimizing bacteremias in the perioperative period has yet to be defined clearly. However, they are clearly beneficial in patients with injuries greater than 60% of body surface and in any other patient in whom the probability of bacteremia with wound manipulation is felt to be high.94 Appropriate perioperative antibiotics are guided by surface cultures. Burn patients are prone to a large number of unusual and often occult infectious complications95 which can result in sepsis and potentially contribute to the development of multiorgan failure. Rapid diagnoses and treatment are facilitated by a high index of suspicion.
Intravascular infections such as suppurative thrombophlebitis and endocarditis typically present in burn patients with fever and bacteremia without localizing signs. Burn patients with endocarditis develop a new murmur in only 9% of cases,96 and only 10% have been reported to present antemortem.97 Of those with septic thrombophlebitis, 68% have no localizing signs and present with fever and positive blood cultures only.98 The diagnosis is made in patients without localizing signs only by thorough examination of all sites of prior cannulation, with surgical exposure of any suspicious sites and complete excision of any involved veins.99 Vigilant care and scheduled replacement of intravascular devices will minimize the occurrence of catheter-related sepsis. Occult intracompartmental sepsis can also present with fever and bacteremia without localizing signs, and is diagnosed only by careful examination and exploration of suspicious compartments.100
Pneumonia, seen in approximately 35% of patients with inhalation injury, adds between 20 and 60% to the expected mortality of burn patients.101 Although a difficult diagnosis to make in critically ill patients, pneumonia should be vigilantly anticipated and aggressively treated with appropriate endobronchial toilet and specific antibiotics. The incidence of nosocomial pneumonia increases with longer durations of intubation,102 emphasizing the importance of judicious use of mechanical ventilation.
Suppurative sinusitis is being recognized with increasing frequency in the intensive care unit, and may be more frequent in patients who are nasotracheally intubated.103 Diagnosis may require examination and culture of material obtained by antral puncture, in addition to plain radiographs and computed tomography. Although there is some controversy about the role of the nasotracheal tube in causing sinusitis,104 treatment involves replacing nasotracheal devices with topical decongestants and appropriate antibiotics. Surgical drainage is reserved for recalcitrant cases.
Acalculous cholecystitis often presents with generalized sepsis without localizing signs in the burn patient, and, like intravascular infection, is a very difficult diagnosis to make.105 Recently, bedside placement of percutaneous cholecystostomy tubes under ultrasonic guidance has become an option in the management of suspected cholecystitis in critically ill patients.106 This technique allows an accurate diagnosis to be made and the condition to be temporarily treated in patients too unstable for immediate operation.
Support of the gut
Bacteria and their products released when the gut barrier fails may fuel the multiorgan failure syndrome. Gut mucosal integrity suffers when mesenteric flow is inadequate and gut blood flow is decreased after burn injury, a response exacerbated by the release of TXA2.107 Thus, support of splanchnic blood flow is an important aspect of multisystem organ failure prevention,108 and this is best done by careful attention to whole-body hemodynamics. There is no substitute for a carefully monitored burn resuscitation.
The enterocyte may be better supported by intraluminal, rather than parenteral feedings, as gut deprived of intraluminal feedings develops mucosal atrophy.109 Early enteral feedings are tolerated in the burn patients110 and may reduce the magnitude of the hypermetabolic response to the injury.111 Parenteral feedings do not prevent gut mucosal atrophy as well as isocaloric and isonitrogenous intraluminal feeds,112 although convincing human data are not available to support the clinical significance of this point.
The value of specific nutrients to support the enterocyte is far less clear than the value of providing adequate mesenteric blood flow and perhaps intraluminal nutrition. However, this is an exciting and active area of research. Glutamine, a non-essential amino acid, is the preferred fuel of the small bowel enterocyte113 as well as other rapidly dividing cells. Sepsis has been shown to decrease glutamine uptake by the small bowel enterocyte, which may result in barrier failure,114 and the addition of glutamine to the nutritional regimen has been theorized to improve barrier function. Glutamine is not a component of commercial parenteral nutritional formulas because of its short shelf life, although the dipeptide is well tolerated parenterally, and has a longer shelf-life.115 Although supplemental glutamine may improve protein balance in surgical patients,115 and may partially reverse gut atrophy,116
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


