ABSTRACT
In this chapter, we review the profound implications that the rapidly emerging field of epigenetics holds for the cosmetic scientist, beginning with an explanation of basic epigenetic principles followed by the role epigenetics plays in the aging process, the impact nutraceuticals and gene-regulating ingredients have on the aging process, and the significant contributions epigenetics can make towards the future of next-generation skin care.
5.6.1 The Human Genome Project Gives Birth To The Epigenetic Revolution
5.6.3 Two Primary Epigenetic Mechanisms
2. Chromatin remodeling and histone modification
5.6.4 Epigenetic Links To Aging
5.6.5 Epigenetics And Aging Skin
5.6.6 Epigenetics Mechanisms In DNA Damage and Repair
5.6.7 Cosmetic Ingredients As Epigenetic Modifiers
5.6.8 Nutriepigenetics: How Diet Alters the Epigenome
5.6.9 Epigenetics: The Unifying Theory Of Aging?
INTRODUCTION
Our understanding that genes can be switched on and off is rather new to geneticists, but the finding that gene expression occurs in response to an immense variety of environmental signals—a phenomenon known as “epigenetics”—is currently revolutionizing all sciences where gene expression comes into play. Medicine, gerontology, botany, toxicology, nutrition, psychology, dermatology, and the cosmetic and personal care sciences are all on the verge of being transformed by discoveries in epigenetics. With over 100,000 papers published between 1992 and 2011, epigenetics is emerging as a paradigm-shifting, rapidly developing branch of genetics that is revolutionizing the way biologists view gene regulation, protein expression, and noncoding DNA that was once thought of as “junk” DNA.
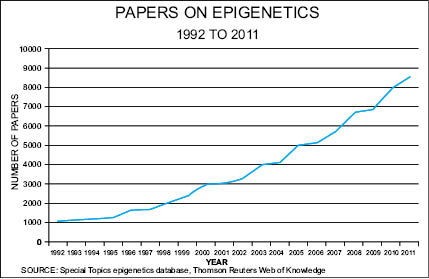
Epigenetics explains how plants know when to bloom, why a gentle touch results in deep relaxation, how cancers form, and how cancer types can be passed from a grandparent to a grandchild. Epigenetics also offers an explanation as to why identical twins don’t always share the same diseases or show signs of aging at the same rate, and why a skin cell looks and acts differently than a brain, bone, or muscle cell. This difference occurs even though they all start from one cell, the zygote—the first cell of the body. More common than gene mutations, epigenetic aberrations that occur in response to environmental signals are being linked to an array of diseases and disorders. These include diabetes, asthma, obesity, Progeria, lupus, rheumatoid and osteoarthritis, metabolic disorder, cardiovascular disease, Down’s Syndrome, Parkinson’s disease, Alzheimer’s, autism, schizophrenia, bipolarism, depression, anxiety, addiction, post-traumatic stress disorder, inflammation, immunosuppression, various symptomologies of aging, and all types of cancers, to name just a few on a quickly expanding list.
In this chapter, the fundamental concepts of epigenetics are described, followed by issues of concern to cosmetic and personal care scientists. These issues include a basic explanation of epigenetics and its mechanisms, how these mechanisms shift with age, and the possible positive influences nutraceuticals and gene-regulating ingredients may have in skin care. For anyone developing next-generation skin care, familiarity with basic epigenetic concepts will prove vital in the future.
5.6.1 THE HUMAN GENOME PROJECT GIVES
BIRTH TO THE EPIGENETIC REVOLUTION
Exploration of the genome—the hereditary DNA sequences of protein-coding genes and noncoding DNA required to make an organism—consumed most of the second half of twentieth-century genetics. At the end of the century an international team of researchers began the Human Genome Project (HGP) with the ultimate goal of mapping and understanding all of the genes required to make and operate a human. Before the project’s completion in 2003, conventional wisdom held that all genetic functions and heredity were specified by the information encoded in an individual’s DNA sequence—the pattern of four nucleotide base pairs (cytosine-guanine, thymine-adenine) from which the three billion rungs on the 23 pairs of the twisted, ladder-shaped set of human chromosomes are formed. All genes are made up of segments of these base pairs arranged in different ways and in different lengths. Acquired changes in cell behavior, including alterations that lead to aging and disease, were thought to be due to genetic mutations, described as unrepaired damage to the DNA sequence accumulated over time. But this turned out not to be true, for one of the gifts of the HGP was the discovery that DNA mutations are not as common as originally believed. It is now known that 1) too few mutations exist to account for the wide variety of human diseases, and 2) the ability of the environment to influence or promote disease and aging does not generally involve mutations (1).
Because of the vast complexity of the human body, by the end of the HGP geneticists expected to identify between 50,000 and 120,000 genes, each one featuring the code for a different protein. Yet a modest 20,500 genes were discovered (2), amounting to less than 2 percent of the entire human genome. Unfortunately, and in view of the early information available, the remaining 98 percent of the genome was cast off as “junk DNA” because not much was known about what it did or why it existed. Later, researchers would find that among this 98 percent were sequences preceding, following, and even in the middle of genes—sequences that govern when, where, and how much of a gene is expressed. Even more intriguing than how little of the genome was devoted to genes, researchers learned that one gene can hold the code for up to 1,000 proteins, making the estimated number of possible proteins produced in the human body between 250,000 to one million (3).
How was it possible that so few genes can produce so many proteins yet only a relatively few proteins are produced at any given time? It seemed obvious that something was happening at the genetic level to direct protein production, but what it was had yet to be unveiled. This “something” could also explain how more than 220 cell types, each type with its own agenda designed to service the tissue where it is found, contain the same genome passed down from the body’s original cell, the zygote. Questions began to arise among geneticists, such as, “If genes are the same in all cells, what causes DNA to be expressed differently? What causes cell differentiation from the zygote to various types of stem cells to, finally, terminal cells unique to specific tissues? And what causes 98 percent of genes to shut down after the gene is no longer needed by the cell?” Those studying aging and disease asked how twins, with exactly the same genome, age at different rates and develop different diseases? Along the same lines, cosmetic and personal care scientists might ask, “How can one person respond to a new ingredient or product with astounding results while others show no response?” None of this can be explained by identifying the 20,500 genes in the human genome.
So what is the answer to all of the questions posed above? The answer, given the present state of knowledge, is that epigenetic chemical modifiers (aka: epigenetic tags), collectively known as the epigenome due to their placement “on” or “above” (“epi”) the genome, are now known to be responsible for gene expression and repression. These modifications occur throughout all stages of an organism’s development, in response to environmental factors such as exposure to toxins or chronic stress, and are implicated in diseases including cancer. Once thought to occur only in response to the environment, including the environment surrounding and within an individual cell, it is now known that the epigenome is in place to “run” the genome, playing genes the way that fingers play piano keys.
Even though a key exists and has the potential to sound a musical note, no sound is made until a finger presses down, either hard or softly, to cause a sound to emerge. In the same way, genes exist and have the potential to create proteins but until the epigenome gives the direction to be played, a gene is not expressed and its resulting protein isn’t synthesized. The gene is “silent,” made mute by epigenetic mechanisms. In an adult somatic cell (a body cell as opposed to a cell used for organism reproduction), 98 percent of the genome is silenced (4). If they weren’t, all genes within every cell would be available for expression. This would result in a wild cacophony of proteins that would wreak havoc in every cell in the body because all cells would be the same, expressed at the same time like an orchestra with no maestro. Life as we know it, where groups of unique cells work together to form and operate an organism, would not exist. This is why, where all cells contain the same set of genes, epigenomes vary from cell type to cell type and even from cell to cell. And, while genes take hundreds of generations to evolve in response to their environment, most epigenetic mechanisms respond quickly to environmental changes, sometimes within seconds. These responses can be stopped, and usually are, when the stimulus is no longer present. But some persistent or particularly harmful stimuli like endocrine disruptors, poor nutrition, or severe stress can entrench epigenetic changes for a lifetime or even for generations. Epigenetics is a young science but it is already clear the epigenome learns from its experience.
Epigenetics is a rapidly emerging branch of biology. Epigeneticists study the vast array of molecular mechanisms that affect the gene activity continually taking place inside cell nuclei (5). In broad terms, these mechanisms are involved in deploying the genetic programs required for the many processes operating during the life span of a cell. Among them are cell lineage development, genomic stability, stress response, pathological conditions, and adaptation to the environment (6). More specifically for the purposes of the cosmetic and personal care developer, epigenetic mechanisms are now known to play important roles in stem cell differentiation (7), olfaction (8), hair growth (9) and graying (10), wound healing (11), inflammation (12), hyper- and hypopigmentation (13), endogenous antioxidant synthesis (14), DNA repair (15), aging (16), cell longevity (17), senescence (18), apoptosis (cell death) (19), allergies (20), and skin diseases such as eczema (21), psoriasis (22), vitiligo (23) and, most likely, acne and rosacea.
Epigenetic mechanisms do not alter DNA sequences but they do change the shape and chemical behavior of the molecules that form DNA. These changes alter gene expression, determining when genes are turned up or down or turned on or off completely. Though varied, epigenetic mechanisms are typically stable and reversible while, at the same time, capable of dynamic and wide-ranging fluctuations, able to both respond to and direct changes in gene expression. Core epigenetic operators are chemical tags that attach to the chromosomal complex known as chromatin, a structure composed of DNA, nucleosome, and RNA molecules that will be discussed in more detail later in this chapter. When present, these tags either block the expression of genes or make them available to be read by transcription factors that work with RNA to translate the genetic code into a protein.
Epigenetic mechanisms occur as a response to environmental messaging, either from the macroenvironment of the organism or the microenvironment of the cell. Until recently, only those heritable mechanisms passed between generations of cells and organisms have been considered epigenetic, but transient forms of gene expression, such as histone modification, are now regarded as epigenetic in their effect. In species that experience a diverse range of environmental conditions, whether they are animals, plants, insects, yeasts, or bacteria, epigenetic mechanisms cause key genes required for survival to be expressed differently throughout the population, thus improving the chances for individual survival (24)—and therefore the survival of the species.
5.6.3 TWO PRIMARY EPIGENETIC MECHANISMS
Research over the past few years has focused on two primary molecular mechanisms believed to participate in most epigenetic events:
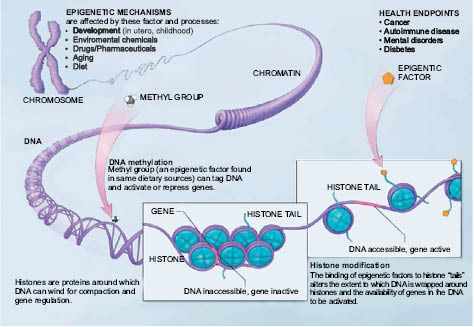
FIGURE 1.4. Epigenetic mechanisms are affected by several factors and processes including development in utero and in childhood, environmental chemicals, drugs and pharmaceuticals, aging, diet, and certain cosmetic ingredients. DNA methylation is what occurs when methyl groups tag DNA and activate or repress genes. Histones are proteins around which DNA can wind for compaction and gene regulation. Histone modification occurs when the binding of epigenetic factors to histone “tails” alters the extent to which DNA is wrapped around histones and the availability of genes in the DNA to be activated. All of these factors and processes can have an effect on people’s health and well-being.
LICENSING: National Institutes of Health. This work is in the public domain in the United States because it is a work prepared by an officer or employee of the United States Government as part of that person’s official duties under the terms of Title 17, Chapter 1, Section 105 of the US Code.
1) DNA methylation and 2) chromatin remodeling, which includes histone modification. Others, such as those mechanisms utilizing several types of small noncoding RNAs (ncRNA) called miRNA, siRNA, and piRNA, as well as long noncoding RNA (lncRNA) (25), are not as well characterized at this time although they are evolving areas of investigative focus.
All epigenetic mechanisms work either singly or, more often, collaboratively in systems of crosstalk that direct cellular development and differentiation in embryonic cells, stem cells, and somatic cells. These mechanisms act as a cell’s memory, allowing it to respond and adapt to its environment, ensuring it knows where it belongs as well as where it has been, the proteins it has produced (or not produced), the threats it has encountered, and how it survived. Further, epigenetic mechanisms determine when a cell should reproduce, how long it should live, and when it should die. In short, epigenetic mechanisms control how, when, how much or how little, and under what circumstances a cell’s genes are expressed. Because of these feats, the epigenome, the totality of a cell’s epigenetic memories, is, quite literally, the “master” of the genome.
The first epigenetic mechanism, discovered almost 100 years ago (26), DNA methylation is also the most researched and understood of the two mechanisms discussed here. It is thought to be the missing, and perhaps main, link between genetics, aging, disease, and the environment (27). Distinct patterns of DNA methylation allow stem cells to differentiate and populate specific tissue types. DNA methylation patterns are known to change with age and are unique to specific disease states. As with single nucleotide polymorphisms (SNPs), which are small deviations in the base pairs that form the rungs on the DNA ladder, identifying DNA methylation patterns and understanding the impact of alterations in these patterns promises to significantly advance our ability to control and affect the aging process and diagnose and treat human disease.
DNA methylation occurs when specialized enzymes known as DNA methyltransferases (DNMTs) add a methyl group to a cytosine molecule that makes up one half of a rung on the DNA ladder. S-adenosyl methionine (SAM) acts as the methyl donor for most if not all DMNT activities and will be discussed later in this chapter.
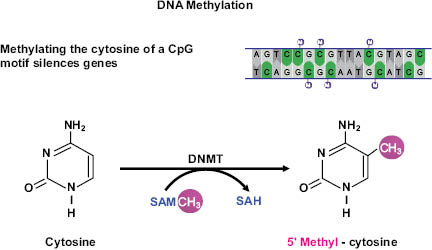
Figure 1.5. Schematic representation of DNA methylation, which converts cytosine to 5’methyl-cytosine via the actions of DNA methyltransferase (DNMT). DNA methylation typically occurs at cytosines that are followed by a guanine (i.e., CpG motifs).
NOTES: SAM = S-adenosylmethionine; SAH = S-adenosylhomocysteine.
Three active DNMTs have been identified in mammals: DNMT1, DNMT3A, and DNMT3B. All three are involved in de novo (new) placement of methyl groups on cytosines, especially in early embryo development where they set up the pattern of methylation that will follow into adulthood (28). DNMTs are also vital for establishing DNA methylation after exposure to methyl donors from the environment. In addition to de novo methylation, the maintenance methyltransferase, DNMT1, the most abundant of the DNMT family, adds methyl groups during cell division to corresponding DNA when one strand is already methylated. This process works throughout the life of the organism to ensure methylation patterns are passed from mother to daughter cells during replication (29).
The addition of the methyl group alters the shape of the cytosine to such an extent that it is invisible when a transcription protein seeks it out during DNA transcription, the process that occurs when sections of DNA are copied onto RNA. If located in an area vital to gene transcription, or if an area features numerous methylated cytosines (known as hypermethylation), the entire gene can be shut down. This process occurs almost exclusively on CpG regions (cytosine linked by one phosphate to a guanine that makes up the other side of the rung on the DNA ladder). CpG regions tend to cluster in CpG islands located near gene-transcription start sites known as promoter regions. In normal tissues, less than 3 percent of promoter CpG islands are methylated (30), which causes the associated gene to be silenced. Approximately 80 percent of the remaining CpG sites, though sparsely scattered throughout the genome, are hypermethylated (31), a cumulative process that becomes more prevalent with each successive level of stem cell differentiation when genes that are no longer needed are shut down.
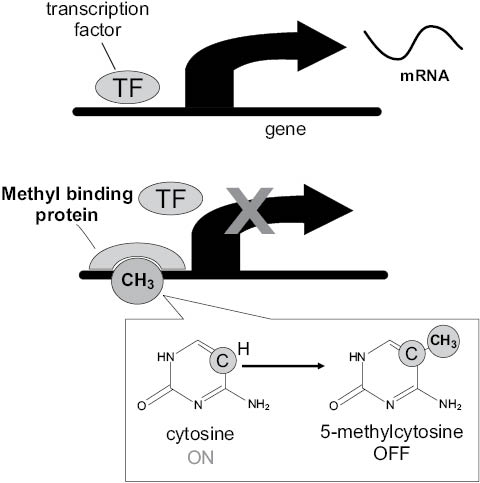
Figure 1.6. DNA methylation can silence gene expression. When cytosines in gene promoter regions are unmethylated (image 1), transcription factors (TF) can initiate the copying of a gene to mRNA. When cytosines are methylated (attachment of CH3) (image 2), access to the gene promoter region is hidden from the transcription factor. Transcription of mRNA does not occur. The gene is silenced.
During aging, this pattern is gradually reversed, resulting in sporadic but higher incidences of methylation in CpG islands, especially those upstream of developmental genes (32), housekeeping genes, and genes involved with cell cycle regulation, tumor-cell invasion, apoptosis, metabolism, cell signaling, and DNA repair (33). These age-related changes are accompanied by a global loss of methylation (hypomethylation) in non-island CpG sites (34), mainly in genes with multiple copies (35); (36). Indeed, global hypomethylation appears to be the most important feature of aging cells and tissues (37). Certain age-associated DNA methylation signatures are so consistent across multiple tissue types they provide an almost perfect correlation of 0.96 with chronological age, making them remarkably accurate calculators of cellular age (38); (39). When measured by correlation, determination of DNA methylation age is about 80 percent more accurate than the telomere length-based clock currently used to determine cellular age. Software intended to calculate DNA methylation age can be accessed online. If DNA methylation proves to be the epigenetic clock of the aging process, this software may be used to identify critical biomarkers for evaluating rejuvenating interventions, including those developed for use in personal care.
Moving beyond the aging process, a broad array of age-related diseases, such as those of an autoimmune, neurodegenerative, or cardiovascular nature, are associated with abberations in established DNA methylation patterns. These aberrations are even more widespread during carcinogenesis, causing deviant DNA methylation to be a major contributor in tumorigenesis by shutting down tumor suppressor genes. Investigation has now shown these genes are turned off in almost all cancers (40), giving cancer researchers a target for new therapies that avoid the devastating side effects of chemotherapy and radiation. Cancer drugs such as 5-Aza-2’-deoxycytidine have been developed to degrade methyl-attaching DNMTs after they are formed, thereby inhibiting the enzyme’s ability to shut down tumor suppressor genes. Unfortunately, these drugs must be incorporated into the genome of the cell, which can cause epigenetic mutations to be passed to surviving daughter cells (41) if the cell does not die. Newer drugs that prevent the synthesis of DNMT1 are currently being developed, but at this time it is not clear if targeting DNMT1 alone will be sufficient to reactivate tumor suppressor genes. If production of DNMT1 is shut down, concerns arise regarding the appearance of unwanted and widespread effects in other areas of the genome where DNMT1 is needed. Only thorough, long-term, and careful examination of these effects can clarify the answers to these concerns. This caution is valid not only for cancer therapies, it holds true for personal care applications as well.
At the time this chapter is being written, researchers at the Cancer Science Institute of Singapore and the Harvard Stem Cell Institute are announcing they have found a novel ncRNA that binds to DNMT1 and prevents DNA methylation of the CEBPA tumor suppressor gene associated with acute myeloid leukemia, lung cancer, and other types of cancer (42). This is the first demonstration of such an effect and is based on the team’s earlier findings that certain RNAs inhibit methylation.
Because aberrations in DNA methylation have dire consequences, including aging, predisposition to cancers, and other diseases passed from one generation of organisms to the next, efforts like the Human Genome Project are currently underway to identify all areas of DNA methylation in the human genome, collectively known as the human methylome. Teams around the world with interests in specific cell types, tissues, diseases, and disorders are also identifying methylome traits unique to their subject of specialty. It is hoped that as these efforts come to fruition, we will be better able to direct the biochemistry that corrects methylome aberrations found in aging, skin disorders, cancers, and diseases of all types, including those induced by ecotoxicants and exposure to aggressive agents such as UV, smog, pesticides, and tobacco smoke.
2. Chromatin remodeling and histone modification
Chromatin is composed of three main molecules:
- a) DNA, the structure that forms the genome;
- b) Nucleosomes, clusters of eight histone proteins around which DNA is wound like thread onto a spool; and
- c) RNAs that first receive a transcribed version of genetic DNA and then translate the DNA code into a protein. Another function of some RNAs may be to identify where and how densely epigenetic tags are placed on chromatin (43), (44).
Chromatin exists in two states of condensation.
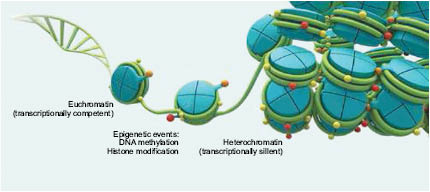
Figure 1.7. Chromatin is found in two states of compaction: euchromatin (transcriptionally competent) and heterochromatin (transcriptionally silent). Epigenetic events such as DNA methylation and histone modification may alter chromatin between these states. Copyright © 2014, American Biotechnologist
Genes located in loosely condensed or “open” regions known as euchromatin are easily accessed by gene-reading proteins called transcription factors and are therefore more actively expressed than tighter heterochromatin regions where genes are repressed, or “silenced” from transcription. Changing chromatin’s state of condensation is known as chromatin remodeling.
Tissue-specific variations in euchromatin and heterochromatin gene regions allow for the development of over 220 cell types in the human body, even though all cells in the body feature virtually the same DNA sequences. In terminally differentiated cells, such as keratinocytes, an estimated 98 percent of the genome is maintained in a heterochromatic state, ensuring the cell does not produce proteins that were required farther back in the originating stem cell line.
Telomeric repeat sequences found on the ends of chromosomes are composed of hypermethylated heterochromatin to ensure chromosome stability (45), suggesting epigenetic regulation may be important in telomere maintenance (46), (47). However, it has been observed that telomeric heterochromatin, once thought to be permanently silent, can be transcribed (48) (49), indicating heterochromatin regions are not a static, closed entity (50) as previously thought.
Indeed, it appears chromatin’s structure and underlying biochemistry support properties that are random and dynamic (51). It is now apparent that chromatin is not rigid, but is continuously remodeled and redistributed along the chromatin chain, allowing heterochromatin regions to “breathe.” Sometimes these behaviors counter the aging process and extend the life span of the organism. However, over time, they may contribute to the breakdown of nuclear, cell, and tissue function, advancing the aging process and age-associated diseases (52).
Histone modifications are a more complex type of epigenetic mechanism than DNA methylation. Not only do histones structurally organize DNA when they form the protein spools called nucleosomes, but they are also subjected to modifications that alter transcription protein binding sites, DNA methylation, and DNA damage responses (53). These modifications usually occur on amino-terminal tails that extend from each histone in the spool.
First discovered in the mid-1990s, some histone modifications, including phosphorylation, sumoylation, and ubiquitination, are transient in that they are associated only with temporary changes in gene expression. Others, such as acetylation and methylation, are more stable. In fact, histone methylation patterns can survive from one cell generation to the next and from organism to organism (54).
Following is a list of well-known histone modifications, the enzymes responsible, and their effects on gene expression.
Acetylation. Histone acetyltransferase (HAT) brings about histone acetylation, causing DNA “threads” to become loosely wound around nucleosome “spools.” This relaxes chromatin into the open formation of euchromatin, making DNA available for gene expression and transcription into proteins. When histones are deacetylated by histone deacetylases (HDACs), chromatin tightens into heterochromatin, increasing the likelihood that genes in this region are silenced and unavailable for transcription into proteins. At least 18 HDACs have been identified in humans (55). The most well known and broadly studied are the seven members of the sirtuin family, especially SIRT1. When gene expression is altered because of an imbalance between HAT and HDAC activity, progression in aging and age-related diseases occur (56). Transcription proteins known to be modified by sirtuins include the cell cycle regulatory gene, p53, the apoptosis-regulating FOXO genes, of which FOXO3a is thought to play a role in protection from oxidative stress, and the TATA-box binding protein TAF1 (57) (58), which plays a central role in mediating promoter responses to various gene activators and repressors. As a result of this activity, sirtuins most likely are involved in modulating cell survival under stress and cellular senescence (59) among other age-related processes.
Methylation. Histone methyltransferases (HMTs) add methyl groups to histones. Histone methylation varies throughout the nucleosome (60), resulting in gene expression or repression depending on the position and number of methyl groups on individual histones (61). At one time histone methylation was thought to be irreversible (62) as these sites are passed between cell generations, but discovery of such enzymes as peptidylarginine deiminase 4 (PAD4) (63), lysine-specific demethylase 1 (LSD1) (64), and Jumonji domain-containing hydroxylases (JHDMs or JMJDs) (65) has proven, at least in some instances, histone methylation is reversible.
Phosphorylation. Histone phosphorylation is critical in gene transcription, chromosome condensation (formation of heterochromatin), DNA repair, and cell death (66).
Ubiquitination and sumoylation. Structurally similar, the role of these proteins is not well understood. Sumoylation is most likely linked to repressing gene transcription while ubiquitination can assist in activating or repressing gene transcription depending upon where in the nucleosome it is associated (67).
DNMTs and histone-modifying enzymes work jointly with chromatin remodeling factors, such as the gene-silencing Polycomb-group proteins (PcG), cyclin-dependent kinases (CDKs) (68), and the Nucleosome Remodeling Deacetylase (NuRD) complex (69), a group of associated proteins with both histone deacetylase and ATP-dependent chromatin remodeling actions that are diminished with age.
5.6.4 EPIGENETIC LINKS TO AGING
Aging is a complex process controlled by environmental and genetic factors (70). These factors lead to genomic instability and epigenetic destabilization and modifications that ultimately throw the organism out of balance (homeostasis), resulting in a compromised stress response and an elevated risk of disease (71). Indeed, it appears that epigenetic changes, more than genetic mutations, drive aging and disease in the second half of life.
Because of this, the epigenetic basis of aging is becoming one of the most popular topics for exploration by researchers concerned with finding keys to reversing the aging process and extending human longevity. Among the most common research topics are the epigenetic links to known or suspected contributory factors to the aging process. These include, but are not limited to:
- • Oxidative damage (ROS, RNS, RCS) (72)
- • Shortened and dysfunctional telomeres (73)
- • Mutation accumulation in nuclear and mitochondrial DNA (74)
- • Alterations in molecular pathways (75)
- • Mitochondrial dysfunction (76)
- • Altered intercellular communication (77)
- • Decline of stem cell functions and populations (78)
- • Altered differentiation of stem cell dependent self-renewing tissues (79)
- • Cell senescence (80) and apoptosis (81)
- • Alterations in systemic hormonal factors (82)
- • Loss of proteostasis (83)
- • Up-regulation of genes with negative impacts on aging, such as the family of inflammatory NFkB transcription factors (84)
- • Down-regulation of genes with positive contributions towards maintaining “youthful processes,” such as the sirtuin family of longevity genes and the endogenous antioxidant regulator Nrf2 (85)
- • Up-regulation of genes with negative impacts on aging, such as the family of inflammatory NFkB transcription factors (84)
After reviewing this list, it can be construed that aging is due to the decline of elements and activities required to maintain the “processes of youth” while those that advance aging become increasingly more pronounced. Simply stated, during aging everything good goes down and everything bad goes up.
Contributing to our understanding of the epigenetic connection to aging is the recent identification of 150 “long life” variants of 70 genes known as Human Longevity-Associated Genes (LAGs) (86). Believed to be rare in normal individuals, these gene variants are associated with longevity in centenarians, where they have been identified in almost 80 percent of this group. While the long-lived seem to have just as many disease-associated genetic variations as everyone else, epigenetic expression of these LAG variants may somehow cancel or take priority over these variations to compress disease and disability into the last few years of life (87). LAGs have recently been identified in protein interaction networks (PINs)
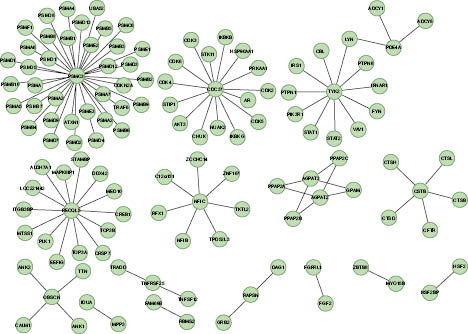
Figure 1.8. Protein Interaction Network (PIN) for Human Longevity-Associated Genes.
Reference: Glessner JT et al. 2013. Copy Number Variations in Alternative Splicing Gene Networks Impact Lifespan. PLoS ONE 8(1): e53846; doi: 10.1371/journal.pone.0053846 where approximately 50 interaction partners have been recognized (88). Although age-associated DNA methylation favors genes with exceptionally low connectivity to other genes (89), when connected to LAGs they become part of an unexpectedly large network of gene communities that play key roles in dictating complex aging processes. Understanding the effects of these various interactions may begin to provide an explanation for how LAGs operate in gene networks and pathways. Further study into the effect epigenetics plays in LAG expression could lead to advances in personalized approaches to longevity and anti-aging.
While beyond the scope of this chapter, it is important to emphasize that virtually all of the molecular mechanisms associated with the aging process are currently under investigation by various researchers to determine links associated with age-related epigenetic modifications. Although research in these areas is just a few years along and, as yet, is poorly understood in its consequences, it is already becoming evident that epigenetic mechanisms are fundamental to all known aging processes and are therefore targets for controlling and even reversing changes in gene expression that occur with age (90). For, unlike genetic mutations, epigenetic mechanisms appear to be reversible and, as a result, may prove to be receptive to environmental influences provided by personal care ingredients. Therefore, cosmetic and personal care formulations seeking to maintain youthful epigenetic patterns while working to reverse the patterns common in aging cells should deliver greater benefits than products of the past.
5.6.5 EPIGENETICS AND AGING SKIN
Epigenetic modifications at specific CpG sites support the fact that aging is coordinated in specific cell groups at different rates and in different ways depending upon the tissue (91). Skin is a particularly suitable model for epigenetic study due to its prolonged environmental exposure and relative ease to biopsy. Upon close examination, skin cells appear to share a high degree of epigenetic uniformity (92).
Changes due to epigenetic modifications acquired with age, a process known as epigenetic drift (93), are also readily apparent. Typical changes in aging skin include fine lines and wrinkling, dehydration, barrier damage, loss of elasticity, dermal thinning, loss of volume around the mouth and above the brows, hyperpigmentation, and age spots. Exacerbated by UV, tobacco smoke, and other environmental elements, these characteristics are the result of decreased epidermal thickness, disorganized corneocytes, depletion of stem cells in epidermal niches, lower levels of collagen types throughout the epidermal-dermal junction and demis, changes in vascularity, reduction in Langerhans cells, thus impacting the immune system, and melanocyte populations impacting pigmentation, typically by overexpression of pigmentation and lipofuscin (94), (95). Given the connection between the environment and skin aging, it is only a matter of time before the epigenetic mechanisms underlying the biological events specific to aging skin are disclosed.
Because Monozygotic (MZ) twins share identical DNA, they are excellent models to determine the environment’s impact on the human epigenome, especially when comparing age-related epigenetic alterations in skin. Several well-known photographic studies showing comparisons of DNA methylation patterns have been conducted on these individuals to exemplify the dramatic visible differences that occur when epigenetic drift is exacerbated by exposure to UV, smoking, stress, diet, and alcohol use.
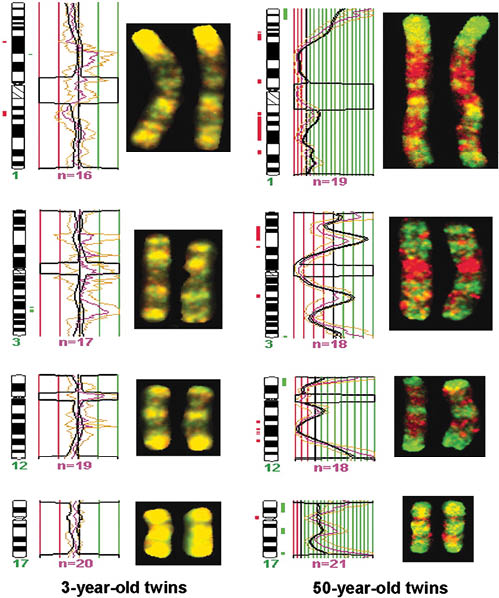
Figure 1.9. Methylome comparisons between 3-year-old and 50-year-old identical twins at chromosomes 1, 3, 12, and 17. Yellow bands note similar distribution of DNA methylation, as indicated in the 3-year-old methylomes, while significant changes in the 50-year-old methylomes are seen by the presence of hypermethylation (green) and hypomethylation (red).
©2005 by National Academy of Science

Figure 1.10. Epigenetic drift in young twins. Changes in gene expression caused by epigenetic drift can begin early in life. These identical twins not only differ in height, but one has asthma while the other does not.
© Reuters. See http://www.thestar.com.my/story.aspx
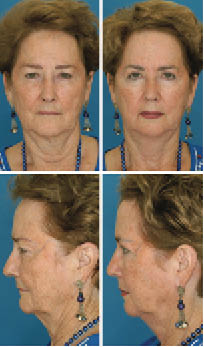
Figure 1.11. Effects of hormone replacement therapy. Twins, age 71. Twin B had 22 more years of hormone replacement therapy than twin A, resulting in a 1.2 lower BMI among other visual differences. Perceived age difference: 7.25 years. Study by Dr. Bahman Guyuron, Department of Plastic Surgery, University Hospitals Case Medical Center.
Copyright ©2009 by the American Society of Plastic Surgeons
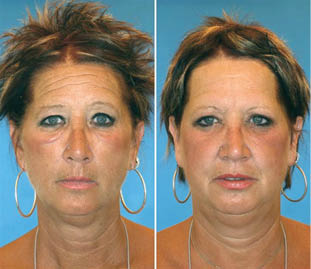
Figure 1.12. Effects of smoking and suntanning. Twins, age 52, Ontario, Canada. Twin A has smoked half a pack a day for 14 years and has 7 times more sun exposure (8-10 weeks a year for 30 years) than her twin. Perceived age difference: 6.25 years. Study by Dr. Bahman Guyuron, Department of Plastic Surgery, University Hospitals Case Medical Center.
Copyright ©2009 by the American Society of Plastic Surgeons

Figure 1.13 Effects of smoking. Twin A has never smoked while Twin B has smoked for 29 years. Note the loose, baggy skin around her eyes, the hollow cheeks, forehead lines, large pores, and lines around her lips. Study by Dr. Bahman Guyuron, Department of Plastic Surgery, University Hospitals Case Medical Center.
Copyright ©2009 by the American Society of Plastic Surgeons
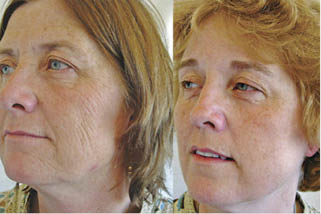
Figure 1.14. Effects of smoking. This twin pair is unique among twins. Not only did they spend their first two decades together but spent their adult years at the same type of job and lived in the same latitude, so their sun-exposure history is well-matched. However, the marked variation between them is that Twin A smoked approximately 52.5 packs of cigarettes a year while Twin B never smoked.
Doshi DN, Hanneman KK, Cooper KD. Smoking and skin aging in identical twins. Arch Dermatol. 2007 Dec;143(12):1543-6.

Figure 1.15. Effects of smoking. Twins, age not available. Both twins smoke; however, Twin B smoked for 14 years longer, as seen by more aging of facial features, including undereye bags, more sagging of the upper eyelids, stronger lines around the mouth, on the chin and cheeks, larger pore size, and more hair loss at the hairline. Study by Dr. Bahman Guyuron, Department of Plastic Surgery, University Hospitals Case Medical Center.
Copyright ©2009 by the American Society of Plastic Surgeons
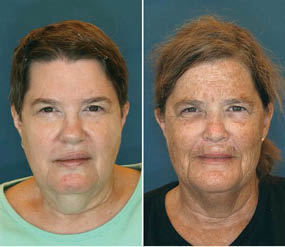
Figure 1.16. Effects of smoking, suntanning and lower weight.
Related posts:
 – THE CELLULAR WATER PRINCIPLE
– THE CELLULAR WATER PRINCIPLE
 – BIOACTIVES GREEN AND SUSTAINABLE INGREDIENTS FROM BIOTRANSFORMATION AND BIOFERMENTATION
– BIOACTIVES GREEN AND SUSTAINABLE INGREDIENTS FROM BIOTRANSFORMATION AND BIOFERMENTATION
 – PEPTIDES FOR ANTI-AGING SKIN CARE
– PEPTIDES FOR ANTI-AGING SKIN CARE
 – CHRONOBIOLOGY OF THE SKIN SKIN CIRCADIAN RHYTHM AND CLOCK GENES: A NEW APPROACH TO SLOWING DOWN THE AGING PROCESS
– CHRONOBIOLOGY OF THE SKIN SKIN CIRCADIAN RHYTHM AND CLOCK GENES: A NEW APPROACH TO SLOWING DOWN THE AGING PROCESS
 – SKIN WHITENER INGREDIENTS
– SKIN WHITENER INGREDIENTS
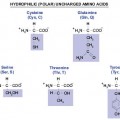
 – AMINO ACIDS
– AMINO ACIDS
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


