This article describes the clinical services for EB in Australia and New Zealand. The history and epidemiology of EB in Australia is described. Current treatment and research achievements are described.
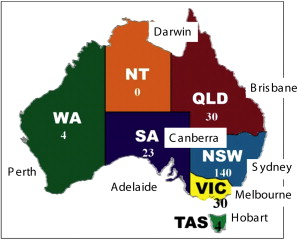
Australia has a population of about 24 million people, and New Zealand, separated from Australia by a 2-hour flight, and consists of the North Island and South Island, has about 5 million people. Australia is a larger land mass than the continental United States, and 90% of its population are concentrated in eight major cities, all of which, except the capital, Canberra, are on the coast. These are the capital cities of the former territories/colonies of the United Kingdom, which only federated into one nation in 1901. Hence, managing patients with an orphan disease presents particular difficulties of distance and tradition. The largest populations are Sydney, New South Wales (4 million); Melbourne, Victoria (3 million); Perth, Western Australia; Brisbane, Queensland; Adelaide, South Australia; Canberra, Australian Capital Territory; Hobart, Tasmania; and Darwin, Northern Territory ( Fig. 1 ).
The first epidermolysis bullosa (EB) clinic to be established was in Sydney, by Mark Eisenberg, a general practitioner with a personal interest in EB. He had the support of the late Brien Walder, head of dermatology at Sydney Children’s Hospital (SCH), and Kieran Moran, a pediatrician was brought in as well. In 1996, I was invited by Walder to assist with this clinic as the dermatologist, since I had been taking care of the EB registry patients at Rockefeller University in New York from early 1994. At that stage there, was ad hoc electron microscopy (EM) in different units and no immunofluorescence mapping (IFM) in Australia. I set up a national diagnostic laboratory for IFM and EM for EB based at St George Hospital, Sydney, within the anatomic pathology department. From 1996, the laboratory has received specimens from around Australia and New Zealand and some surrounding Asian countries shipped in Michel’s media. EM is provided by C.W. Chow of Royal Children’s Hospital, Melbourne. In addition, I began seeing adults with EB at St George Hospital and in my part-time private practice, assisted by my practice nurse, Lesley Rhodes.
Since this first EB clinic was established in Sydney, two other EB clinics started, one at Royal Children’s Hospital, Melbourne, run by dermatologists George Varigos, John Su, and David Orchard, and in Adelaide at the Women’s and Children’s Hospital, originally by Julie Wesley and now by Lachlan Warren. The EB clinic at SCH was expanded with the addition of a medical geneticist, Anne Turner, in 2000, and an additional dermatologist, Orli Wargon, in 2003, after the death of Walder, and a part time EB nurse in 2007, Louise Stevens. In addition, physical, occupational, and pain therapists attend the clinic, which is held monthly as well as a social worker. Newborn infants with EB usually are transferred to SCH from other hospitals in the state of New South Wales.
When EB patients are 16 years of age, they transition to St George Hospital, Sydney, another teaching hospital of the University of New South Wales Medical School. Here the more severe patients with recessive dystrophic EB (RDEB) are reviewed every 3 months in the ambulatory care unit, with a full skin check after a bath, possible biopsies, and infusions as needed, while they are reviewed by designated experts in hematology, renal, endocrinology, pain service, gastroenterology, as needed ( Fig. 2 ).
In New Zealand, Nick Burchall established the service for EB, based in Auckland, now assisted by Deanna Purvis, and Dystrophic Epidermolysis Bullosa Research Association (DebRA) New Zealand raised funds for several regional EB nurses who assist with the management of EB patients around the country with him and regional dermatologists.
Australasian EB registry
In 2005, the author established a national EB registry based at St George Hospital. There are currently 242 patients enrolled, 140 of whom are in New South Wales (see Fig. 1 ). According to figures by the DebRA charities in New South Wales, Victoria, and SA and the newly formed DebRA Australia, there are likely to be about 400 patients with more severe forms of EB and probably 1000 or more if milder cases are included. DebRA New Zealand has about 40 members (A Kemble-Welch, personal communication, May 2009), and 11 are currently enrolled in the registry. To be enrolled, patients have to be examined by one of the EB clinic dermatologists and have confirmatory biopsies or genetic testing to confirm the subtype of EB. Currently the prevalence of EB is 10 per million population, but in reality it will be higher than this.
Provision of care
In Australia, the public hospital clinics are all free to patients with citizenship or permanent resident status. What is not covered is the cost of the expensive dressings, which have been supplied ad hoc by some local hospitals/charities or DebRA. The registry has enabled the author and colleagues in conjunction with DebRA Australia to secure funding for the dressings for the next 4 years for EB patients, up to $16 million. Caregivers allowances are available for the more severe patients, but home nursing is difficult to obtain in Australia.
Provision of care
In Australia, the public hospital clinics are all free to patients with citizenship or permanent resident status. What is not covered is the cost of the expensive dressings, which have been supplied ad hoc by some local hospitals/charities or DebRA. The registry has enabled the author and colleagues in conjunction with DebRA Australia to secure funding for the dressings for the next 4 years for EB patients, up to $16 million. Caregivers allowances are available for the more severe patients, but home nursing is difficult to obtain in Australia.
Research and teaching about EB
The author’s research has been performed at St George Hospital ( Fig. 3 ) and has focused on genotype to phenotype correlations in EB, skin cancers in EB, and novel therapies for EB. Regularly there are organized seminars and lectures at major national and international congresses and invited lectures about EB to improve awareness.
Dystrophic EB
In RDEB, the author and colleagues have discovered nine novel COL7A1 mutations and reviewed all collagen VII mutations causing dystrophic EB DEB. In dominant dystrophic EB (DDEB), the most common mutation in Australia is G2043R, and the author and colleagues have published the first prenatal diagnosis case in DDEB and the first Aboriginal family with DDEB and novel mutations underlying EB pruriginosa. The author and colleagues attempted to use the baculovirus system to introduce the COL7A1 gene, which was able to incorporate the gene, but it did not transduce into human keratinocytes or fibroblasts.
EB Simplex
A collaborative laboratory at Royal Brisbane Hospital has been confirming keratin 5 and 14 mutations identified in the author’s research laboratory at St George Hospital as well as doing routine screening. One of the author’s patients has had an extremely severe form of Dowling-Meara EBS, and the author and colleagues explained how this had occurred due to the mismatch of the mutated amino acid and its position, Krt14 M119T, with another family with localized EBS, with Krt14 M119V. The author and colleagues discovered a patient with EBS-DM caused by a unique deletion in Keratin 5 and also patients with codominant inheritance of Krt5 and Krt14 mutations. Altogether, there are at least 10 novel keratin 5 or 14 mutations in the St George Hospital patient population with EBS. The author and colleagues reviewed a keratin 14-negative case in the context of all K14-recessive EBS as the 14th reported case in the world. Recently, the author and colleagues had their first case of EBS with late-onset muscular dystrophy caused by a novel plectin mutation and another case from the Philippines referred with autosomal dominant EBS and ankyloblepharon.
Junctional EB
Thanks to collaborators overseas, the author and colleagues have been able to have mutation studies done on the relevant genes, depending on IF mapping findings. Several new integrin B4 and laminin 332 mutations were published in conjunction with the Uitto group. The author and colleagues set up a rapid screening test for the most common LAMB3 mutations seen in Australia, R42X and R635X, and other specific mutations with the help of laboratories overseas, at the laboratory in Brisbane to enable prenatal diagnosis of Herlitz JEB in Australia. Other studies have led to papers on junctional EB (JEB) with pyloric atresia showing that integrin B4 mutations do not always cause this, and the author’s group was the first to find that heterozygote carriers of COL17A1 mutations could have reduced staining with antibodies in their skin, making this a useful test for situations in which, as in this case, the proband already was dead and had not been tested. The author and colleagues also recognized a unique variant of JEB caused by the granulomatous eyelid lesions, and in conjunction with Irwin McLean’s group, were able to show that this patient had a unique mutation in the same exon of LAMA3a that occurs in laryngo-onycho-cutaneous (LOC) syndrome, proving that LOC was a subtype of JEB.
Squamous cell carcinoma
In conjunction with Jack Arbiser, while in New York, expression of angiogenic factor basic FGF was measured in the urine of the author’s patients at Rockefeller. There were extremely high levels in 13 patients, particularly the RDEB patients. This was confirmed in further samples collected. Among the author’s cohort of DEB patients, it has not been found that positive staining with collagen VII antibodies LH7.2 and FNC1 increases their risk of developing SCC, or that negative staining protects them from SCC. The author and colleagues also have contributed to international collaborative studies in SCC with the Marinkovich group at Stanford, California and the South group in Dundee, Scotland.
Related posts:
 How to Take Skin Biopsies for Epidermolysis Bullosa
How to Take Skin Biopsies for Epidermolysis Bullosa
 Epidermolysis Bullosa Simplex with Muscular Dystrophy
Dilated Cardiomyopathy in Epidermolysis Bullosa
Bone Marrow Stem Cell Therapy for Recessive Dystrophic Epidermolysis Bullosa
Epidermolysis Bullosa Simplex with Muscular Dystrophy
Dilated Cardiomyopathy in Epidermolysis Bullosa
Bone Marrow Stem Cell Therapy for Recessive Dystrophic Epidermolysis Bullosa
 Epidermolysis Bullosa in France: Management in the National Reference Center for Genodermatosis
Epidermolysis Bullosa in Japan
Epidermolysis Bullosa in France: Management in the National Reference Center for Genodermatosis
Epidermolysis Bullosa in Japan
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


