and Emanual Maverakis3
(1)
University of British Columbia, Vancouver, British Columbia, Canada
(2)
New York Medical College, Valhalla, NY, USA
(3)
University of California Davis, Sacramento, CA, USA
2.1 Epidemiology
AE is not specific to any ethnic population, as cases have been reported from all around the world [1]. It is globally widespread with an estimated incidence of 1 in 500,000 children [1–4]. According to the cases reported in the literature, its prevalence seems higher in populations from the Mediterranean basin, probably because of their relatively high overall consanguinity [3]. There is also no gender predilection observed in AE [1, 3]. Compared to the United States, the diagnosis of AE may be more difficult in developing countries where dietary zinc deficiencies are quite common, a problem emphasized in the World Health Report 2002 [5]. Approximately two billion individuals may be zinc deficient in these regions of the world [6], where infants and children are the most affected. The regions particularly affected by zinc deficiency problems include Southeast Asia and sub-Saharan Africa, since about 40 % of their preschool children have been reported to have zinc-related growth problems [7]. It has been reported that moderate zinc deficiency affects approximately 3 % of adolescents in rural areas of Middle East and North Africa [8]. Correcting this situation would have dramatic impacts on the morbidity and mortality of young children and modest effects on their growth. However, it is important to tackle malnutrition of these regions as a whole, instead of undertaking zinc deficiency in isolation. As a result, including zinc in a multiple micronutrient supplementation and promoting their use would be an effective method of dealing with this situation [9].
Even though AE is not as prevalent in the United States as it is in countries with high rates of consanguinity, acquired zinc deficiency has been reported in American newborns and children. There have been several studies that reported of American infants having exceptionally low hair and plasma zinc levels [10]. Deficiency in zinc, may have serious and permanent growth and developmental effects on children and infants of which some were reported in a study from Denver in 1972 where some of the children with zinc deficiency also suffered from growth retardation [11]. The main contributing factors resulting in infant zinc deficiency in the United States include popular infant milk formulae having low zinc concentration and the necessity of large amounts of zinc for infant rapid growth [12].
Lactation, alcoholism, old age, and metabolic disorders are associated with zinc deficiency in the American adult demographic [8]. A recent study found that about 30 % of pregnant women of low socioeconomic status had low body zinc levels. Insufficient maternal zinc levels could have severe effects on prenatal growth and development of the child, such as congenital malformation of the central nervous system [12]. In the light of this, more research and attention should be devoted to increasing knowledge, raising awareness, and correction of these nutritional problems.
2.2 Etiology
2.2.1 Molecular Etiology of AE
Presently, AE is considered a treatable disorder since it can be simply managed with zinc supplementation once diagnosed. As a result of the profound effects this disease has on human physiology, which are particularly pronounced in infants, medical science still considers it as one of the most intriguing and interesting disorders. AE is suggested to have an autosomal recessive mode of inheritance by genealogic data [13].
AE presents a multitude of opportunities for research; however, at present only rare cases are referred to academic medical centers since this disorder is easily treatable. Consequently, the study of this disorder is rarely done. Two main hypotheses are proposed. One is based on a problem of zinc bioavailability and the other on an intrinsic defect of zinc transport by the affected individual.
2.2.2 Hypothetical Etiology 1: The Alteration of Zinc Bioavailability
As patients with AE can obtain only a small amount of zinc through dietary sources, supplementing the diet with sufficient amounts of zinc can raise the level of zinc in blood plasma to a normal level and result in the resolution of the disorder. Although the amount of zinc in breast milk and infant formula are roughly equal, the zinc in breast milk has been observed to be more absorbable by infants with AE and this phenomenon has become an interesting matter among dermatologists, pediatricians, and medical researchers [14].
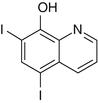
To date, research has indicated that the zinc absorption process is very complex. Research laboratory results illustrate that the zinc binding ligand found in human milk facilitates zinc absorption in the intestine and the ligand’s presence results in high bioavailability of zinc in human milk [15] (Fig. 2.1).
Figure 2.1
Molecular structure of picolinic acid
Evans and Johnson found the concentration of picolinic acid, a zinc binding ligand, in human milk to be much higher than that in either bovine milk or infant formulas. The higher concentration of picolinic acid in human milk could result in the formation of additional zinc picolinate complexes which would be more absorbable by the intestine compared to ionic zinc or zinc complexed with other ligands [15]. Conversely, Rebello et al. found the amount of picolinic acid present in human pancreatic juice or intestine to be less than 2.5 μM. They also found the amount of picolinic acid present in human milk to be approximately 3.7 μM and declared it insignificant in zinc absorption due to its minimal quantity. Rebello et al. also emphasized that they cannot explain the inordinately high values of picolinic acid reported by Evans and Johnson [16]. Later on, it was discovered that supplementation of picolinic acid only increases zinc turnover without increasing retention since its administration to individuals only increased zinc excretion, which would lead to zinc depletion in the absence of adequate zinc supplementation [17, 18] (Fig. 2.2).
Figure 2.2
Molecular structure of zinc picolinate (zinc bound picolinic acid)
Eckhert et al. discovered that alongside elemental zinc, breast milk contains a protein zinc-binding ligand with a low molecular weight of about 10,000 Da, while the ligand alongside elemental zinc observed in bovine milk has a greater molecular weight. The ligand present in bovine milk does not improve zinc absorption; as a result AE symptoms often appear when the infants’ diet is switched to bovine milk [19].
Casey, Walravens, et al. were able to find protein zinc-binding ligands in the duodenum with the same properties of the ligand described above, in pancreatic secretions and they also confirmed that the weight of the ZBL is significant for its function [20]. Recently, researchers have hypothesized that AE may be caused by a mutation in the gene SLC39A4 coding for the hZip4 Zn transporter present in intestinal cells of the intestine [3, 4]. This hypothesis is supported by studies that have concluded that AE patients and healthy individuals have similar duodenal ZBL secretions or that the amount of ZBL present in the intestine is insignificant in zinc absorption [16, 21].
Following the acknowledgment that this zinc deficiency condition is a disorder, the main concern of medical researchers has been the discovery of the mechanism for zinc absorption from a regular diet: a mechanism that seems to function insufficiently in AE patients. Interestingly, Casey et al. determined that zinc absorption was decreased in patients with AE compared to individuals in the control group, even though both groups had the same amount of ZBL present in their duodenal secretion [21]. At the time, it was concluded that ZBL in duodenal secretion of AE patients did not function sufficiently, which could be due to their abnormal nature as a result of the AE disorder [21]. Moreover, an alternative hypothesis is that since zinc absorption was decreased in AE patients, while both groups (AE patients and control group) had similar duodenal ZBL secretions, the insufficient zinc absorption may be due to malfunction of the zinc transporters present in apical intestinal cells, impairing the uptake of zinc [3, 21]. This hypothesis will be discussed later in this section.
Cousins and Smith noticed that only 10 % of the zinc in fat-free breast milk was accompanied by the ZBL that has the low molecular weight; when more zinc was added in vitro to the breast milk, almost all the zinc became ZBL bound [22]. Lonnerdal’s research indicated that an increase in the amount of dietary protein led to an increase in zinc absorption in a linear fashion, which may have been as a result of amino acids released from the protein keeping the zinc in solution and increasing bioavailability of the zinc [23–25]. Amino acids such as histidine and methionine as well as EDTA, a low molecular weight ion, and organic acids have been known to boost zinc absorption and have been used for zinc supplements [23]. For instance, clinical studies have shown histidine to have a positive effect on zinc absorption since it is a good chelator of zinc [26]. It is important to note that high doses of histidine were reported to enhance urinary excretion of zinc, hence the molar ratio of histidine to zinc is of importance when considering the administration of histidine to zinc deficient patients [27]. It was also found in a clinical study that infants who were fed formula with a high protein concentration had lower plasma zinc concentrations than infants fed formula with less protein [28]. On the other hand, zinc found in infant formula was observed to be more absorbable compared to bovine milk, which could be due to the inhibitory effect of casein that is present in bovine milk on zinc absorption [25]. This could be caused by phosphorylated serine and threonine residues on partially undigested casein subunits that bind zinc and reduce zinc bioavailability [29, 30]. Interestingly in a recent study, it was found that casein phosphopeptides (CPP) added to a phytate-containing solution significantly increased calcium and zinc absorption in suckling rat pups as well as in human intestinal cells (Caco-2) in culture [31]. A human study showed that CPP addition to a high phytate meal had no effect on zinc absorption, as a result the effect of CPP may be dependent on the phytate content of the meal [32].
Menard and Cousins have noted that the zinc found in the intestine tends to be bound to citrate, which has a molecular weight of 600–650 Da [33]. This zinc-citrate compound is part of the ZBL mentioned previously; however research has indicated that the presence of citrate as part of the ligand complex does not enhance the absorption of zinc in the rat intestine [34].
Due to the significant interest of medical researchers concerning ZBL, there have been many research reports in this area. However, the exact role of ZBL in zinc absorption is yet to be elucidated. Sang et al. stated that prostaglandin E2 facilitates zinc absorption in the intestine [35]. Furthermore, the total amount of zinc present in the human body was seen to have an effect on its absorption. For instance, in an individual with low levels of zinc, zinc absorption is more widespread and would occur at a higher rate compared to cases where the amount of zinc present in the body was sufficient or more than the required amount [23].
Other factors such as the presence of iron, cadmium, low-molecular-weight ligands, and chelators also affect zinc absorption [23]. For instance, when adult individuals were administered high doses of inorganic iron in solution, there was a decrease in zinc absorption as plasma levels of zinc were seen to diminish [23, 36]. But when the administered solution also contained ZBL, zinc absorption was unaffected. It is likely that the inhibitory effect occurs only when iron-to-zinc ratio is very high and is in solution. When the same amount of high dose iron was administered to adult individuals as supplement tablets, no inhibitory effect on zinc absorption was observed; which suggested that only iron in solution can affect zinc absorption [23]. Interestingly, when infants were fed iron drops (30 mg/drop), zinc absorption was also unaltered [37]. This suggests that iron supplementation does not affect zinc absorption and if iron and zinc supplements are administered apart from meals, an inhibitory effect will be found only when the iron-to-zinc ratio is very high [23].
Furthermore, nontoxic levels of cadmium were seen to have no effect on zinc absorption. Since zinc can form a complex with low-molecular-weight ligands or chelators, such as ZBL, an increase in levels of ligands or chelators were observed to have a positive effect on zinc absorption. Moreover, amino acids like histidine that are good chelators of zinc can increase zinc absorption in the intestine; however, very high levels of histidine often result in excess urinary excretion of zinc and decrease in plasma zinc levels [23].
2.2.3 Hypothetical Etiology 2: A Defective Zinc Transporter
This second hypothesis is supported notably by arguments against a problem of zinc bioavailability. At least two studies had indeed concluded that AE patients and healthy individuals had similar duodenal ZBL secretions or that the amount of ZBL present in the intestine was insignificant in zinc absorption [16, 21]. If the problem in AE patients did not relate to zinc consumption itself, then it could be due to a defect in the molecular protein ensuring its absorption from the intestinal lumen.
Cloning of the first human zinc-specific transporters is quite recent, as ZnT-1 (SLC30A1) and ZnT-2 (SLC30A2) were identified in 1995 [38] and 1966 [39], respectively. In 1997, as they were working on a mouse model of pallid mutant, Huang and Gitschier serendipitously found that homozygous mutations of ZnT-4 (SLC30A4) caused the lethal milk phenotype in the strain they were studying [40]. Interestingly, lethal milk, which is a recessive condition, could be considered as the murine counterpart of human AE, keeping in mind that the consequence of biallelic ZnT-4 mutations are not seen in the mutant mouse herself, but in her suckling offspring. Mutations of ZnT-4 entail a decrease in milk zinc levels of lethal milk mothers, which result in a generalized zinc deficiency and eventually death of the suckling pups.
Yet, SLC30A4 turned out not to have any link with AE, as its involvement in the disease was ruled out by sequencing in AE families [41]. Several other candidates were discarded this way, including ZnT-1 to ZnT-3 (Küry et al., unpublished data) and ZnT-5 (aka hZTL1 or SLC30A5) [42], until Wang et al. highlighted a susceptibility locus for AE on 8q24.3 [43]. Within this locus, two independent teams cloned the gene SLC39A4 [4, 44], presenting a high degree of homology with the ZIP (zinc/iron-like proteins) family of zinc transporters identified several years also in plant and yeast [45–47] and with the three human ZIP family freshly identified [48]. Both teams could establish a relationship between homozygous or compound heterozygous mutations of SLC39A4 and the symptoms of zinc deficiency in several AE patients originating from Tunisia, France, Egypt and Jordania [4, 44]. Since then, the relationship between AE and mutations of SLC39A4 was largely confirmed, as more than 40 mutations were reported worldwide that are distributed along the gene [3, 44, 49]. Recommendations on the molecular diagnosis of AE were detailed in a clinical utility gene card [50].
Apart from likely founder effects noted for instance for the Tunisian mutation c.1224_1228del (p.Gly409Leufs*7) [3], mutations are almost always private and no hotspot is observed within the gene. A slight recurrence can however be noted for a few mutations such as c.599C > T (p.Pro200Leu) or c.192 + 19G > A (p.?). The mutations encountered so far are nonsense, missense and splice site mutations or small frameshift insertions/deletions. No large rearrangement encompassing one exon or more has ever been reported. All genuine cases of AE have either a homozygous SLC39A4 mutation or compound SLC39A4 heterozygous mutations. Beside these cases following a Mendelian autosomal recessive mode of inheritance, transient forms of zinc deficiency have also been observed in heterozygous carriers of SLC39A4 mutations, which would rather follow an autosomal dominant mode of inheritance with incomplete penetrance (Küry et al., unpublished personal observation). The inconsistency of this transient form of zinc deficiency suggests that its occurrence is subjected to the co-existence of an additional risk factors, which might be prematurity, intestinal pathology or a nutritional factor decreasing zinc bioavailability.
According to its function, and its expression-, tissue- and cellular-localizations, SLC39A4 immediately appeared as the ideal candidate for explaining AE. On the one hand, it is highly expressed in the kidney, but most importantly in the duodenal and jejunal parts of the intestinal tract [2, 4], that is to say the main sites of dietary zinc absorption and thereby of human zinc homeostasis. On the other hand, its related protein SLC39A4 (or ZIP4) is localized at the apical membrane of the enterocytes [4], where it transports dietary zinc from the intestinal lumen to the intracellular compartment. Rapidly, functional studies in cellular and murine models showed that SLC39A4 transporter is a key component of a human zinc homeostasis [51] and they confirmed the correlation between SLC39A4 mutations and severe zinc deficiency [52–56]. AE patients’ mutations were shown to either diminish the efficiency of zinc transport by SLC39A4, or to induce its mislocalization in the nuclear envelope and endoplasmic reticulum, which could be attributed to the misfolding of the protein and prevention of its proper glycolysation and localization [51, 57].
The finding of the molecular cause of acrodermatitis enteropathica gave new insight on the mechanism of intestinal zinc absorption. Several studies focused then on SLC39A4, which allowed better comprehension of the overall phenomenon. Thus, the first step for zinc absorption involves the arrival of zinc to the intestine after the food is ingested by the individual and partially digested both mechanically and chemically in the mouth cavity and stomach. Subsequently, as the zinc comes in contact with the apical membrane of the intestinal villous, where the transmembrane protein SLC39A4 (ZIP4) is located, the zinc is actively transported into the enterocyte from the small intestinal lumen [53]. Research on human cells over-expressing the ZIP4 gene showed increased accumulation of 65Zn, indicating a carrier-mediated uptake process [53]. Moreover, in vivo and in vitro studies have shown that SLC39A4 undergoes posttranscriptional regulation in response to zinc levels: in zinc-deficient conditions the SLC39A4 (ZIP4) protein was concentrated on the cell membrane whereas in zinc-replete cells SLC39A4 was sequestered and mainly found in intracellular compartments [51, 53]. Studies on the mouse homologue of SLC39A4, mZIP4, highlighted its control by zinc availability: in zinc deficient conditions, mZIP4 accumulated at the plasma membrane, whereas zinc repletion increased its endocytosis [51, 57]. This mechanism would prevent intracellular accumulation of zinc and thereby from zinc toxicity when dietary zinc amounts are adequate [53]. This responsiveness of SLC39A4 to variations of extracellular zinc conditions is altered by certain AE mutations, which impede the accumulation of the protein during dietary zinc deficiency [57].
2.2.4 Alternative Forms of AE-like Zinc Deficiencies
Interestingly, about half of the patients suspected of having AE have no mutation in coding introns, or promoting sequence of SLC39A4 [58]. In addition, a few patients with a severe form of zinc deficiency very suggestive of AE exhibit only a heterozygous SLC39A4 mutation [58]. This suggests either possible alterations of yet unidentified regulating sequences of the SLC39A4 gene in these patients, or dysregulation of epigenetic mechanisms involving methylation or miRNAs. Alternatively, other zinc transporters might be affected in some cases of acrodermatitis enteropathica [44, 59, 60]. From a semantic point of view, cases of zinc deficiency not caused by homozygous or compound heterozygous mutations of SLC39A4 are not genuine AE and would rather be labelled as AE-like disorders.
So far, only one form of inherited zinc deficiency alternative to AE has been identified in humans. It is caused by mutations of SLC30A2 [61], a member of the very same family as SLC30A4 that is involved in the mouse lethal milk phenotype. Incidentally, the transient form of zinc deficiency associated with SLC30A2 is more similar to mouse lethal milk than to human AE. Mutations of the zinc transporter gene induce decreased milk zinc levels in the nursing mother, which are responsible for a lactogenic transient neonatal zinc deficiency (TNZD; MIM#608118) in the exclusively breast-fed baby. TNZD would be inherited according to either autosomal recessive [61, 62] or autosomal dominant [63–65] mode. Zinc transporter SLC30A2 (or ZNT2), encoded by SLC30A2, contributes to the efflux of zinc from cytosol of secreting mammary epithelial cells to milk, by transporting labile zinc from cytoplasm to endosomal vesicles which is then trafficked to the cell membrane and exocytosed to the milk [66].
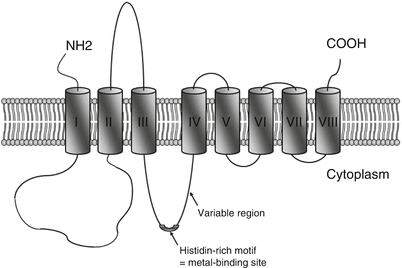
From the examples of AE and TNZD, one could expect other forms of zinc deficiency related to zinc transporters. Beside SLC39A4 and SLC30A2, human zinc homeostasis is indeed sustained by two zinc transporter families of 10 SLC30 (ZNT) and 14 SLC39 (ZIP) members [67–70], the alteration of which might have impact on body zinc levels. Knock-out mouse models showed that ZNT transporters regulate intracellular zinc concentration by either promoting zinc efflux or zinc transport into intracellular vesicles [71]. In contrast, ZIP transporters ensure the uptake of zinc into cells [72]. Members of the ZNT family have six transmembrane domains with a long histidine loop between transmembrane domains IV and V, which is likely a zinc-binding domain [73, 74]. ZIP family members are characterized by eight transmembrane domains organized into two blocks of three and five domains separated by a histidine-rich cytoplasmic metal-binding site, as seen in Fig. 2.3 [45, 75].