Endothelium in Inflammation and Angiogenesis: Introduction
|
Overview of the Vascular System
The blood vascular system is a continuous series of hollow tubes that carry blood from the heart to the tissues and back again. Blood exits the heart through the aorta that gives rise to a series of diverging, progressively narrower muscular arteries and arterioles, delivering blood to all organs of the body. The terminal arterioles arborize into an interconnecting network of microvessels, mostly capillaries that nourish and cleanse the peripheral tissues. The capillary network empties into a series of converging venules and progressively larger veins that ultimately return the blood to the heart. All segments of this vascular system are lined by a one-cell-thick layer of epithelium-like cells called endothelium (Fig. 162-1). All vascular endothelial cells (ECs) share common features and functions, and hence may be collectively described as one cell type. However, ECs from one segment of the vascular system may differ in significant ways from the ECs at other anatomic sites. Blood vessel ECs differ from lymphatic ECs, which are not discussed in this chapter.
Figure 162-1
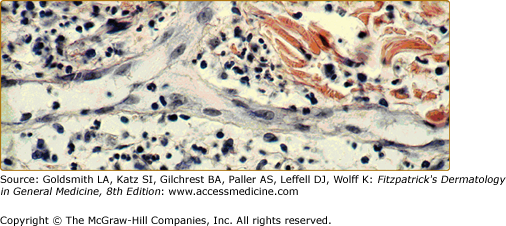
Endothelium. In this postcapillary venule, endothelial cells are seen to line a channel. Nuclei are seen en face and in cross-section, and activated endothelial cells appear to have an edematous cytoplasm, which increases their visibility. This specimen is from an inflammatory lesion (Sweet syndrome; see Chapter 32) without signs of vessel damage.
Blood vessel ECs must perform three important constitutive functions. First, vascular ECs must maintain homeostasis of the blood, which consists of roughly equal volumes of fluid (plasma) and cellular elements (erythrocytes, leukocytes, and platelets). The plasma contains the proteins of the coagulation system. In addition, both leukocytes and platelets have membrane receptors that trigger cellular activation on contact with the extravascular environment. The EC lining prevents intravascular activation both of the coagulation system and of these responsive cellular elements, yet keeps both systems poised respond to wounding or infection.
Second, ECs must separate circulating blood from the tissues. The barrier formed by ECs permits limited passage of fluid and solutes, allowing the blood to nourish the tissues while displaying “permselectivity” for macromolecules and presenting a virtually impenetrable barrier for blood cells (except at specialized sites of high permeability and leukocyte trafficking). In other words, vascular ECs regulate rather than prevent interactions between blood and tissues.
Third, ECs must regulate local blood flow. Because ECs are arranged in alignment with flow, their contraction opens gaps between cells, enhancing permeability without altering flow (see Fig. 162-1). However, ECs indirectly regulate flow by acting on smooth muscle cells (SMCs), which are oriented almost parallel to the vessel’s circumferential or radial axis.
ECs do so principally by the generation of SMC relaxants [e.g., nitric oxide (NO), prostaglandin E2 or prostaglandin I2 (PGI2), and endothelium-derived hyperpolarizing factor] or contractants (e.g., endothelin) or by catalyzing the generation of vasoconstrictors from plasma protein-derived mediators (e.g., angiotensin II through the action of EC-expressed angiotensin-converting enzyme, which also inactivates bradykinin, a plasma protein-derived agonist that acts on ECs to generate NO.
Structure and Function of the Skin Vasculature
Approximately three decades ago Irwin Braverman described the organization of the vascular network of human skin, which he summarized in 1989.1 The human dermal vasculature consists of two interconnected systems—(1) a superficial and (2) a deep vascular plexus—with additional vascular networks surrounding sweat glands and hair follicles2,3 (Fig. 162-2). The superficial vascular plexus (SVP) is comprised of paired arterioles and venules that form an interconnected network of vessels coursing on a plane parallel to and just beneath the epidermal surface.3 Capillaries arise from the arterioles, extend upward within the papillary dermis at sites between the epidermal rete ridges, and then loop back down to the venules forming arcade-like structures. The basement membrane of the arterioles appears homogeneous by electron microscopy whereas that of the venules is multilayered. In normal skin, most of the capillary loop is invested with a basement membrane that resembles that of the arteriole, acquiring a venule-like investiture only at the level of the deepest rete just proximal to the anastomosis with the venule of the SVP.2 There are no ultrastructural differences between capillary loops at different sites of the skin. Inflammatory leukocytes extravasate through the venule-like portions of the capillary and the venule proper but only rarely infiltrate through the arteriole-like portions of the capillary or the arteriole itself.
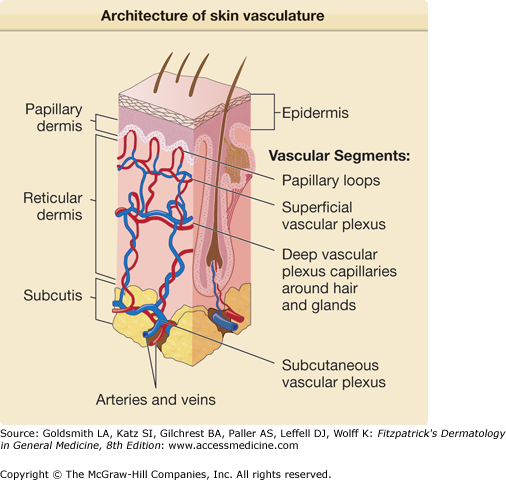
The arterioles and venules of the SVP are connected by short, straight vessels to the arterioles and venules of a deeper planar network of anatomizing vessels called the deep vascular plexus (DVP) (see Fig. 162-2). The plane of the DVP is parallel to that of the SVP and courses above the boundary between the reticular dermis and the underlying subcutis (see Fig. 162-2). The DVP is fed through penetrating vessels from the subcutis. The DVP is drained by valve-containing veins into the subcutaneous fat. The vessel wall in the SVP consists of discontinuous layers of elastic fibers and SMCs. The DVP contains arterioles and venules of larger caliber consisting of three layers: (1) an intima, (2) a media, and (3) an adventitia. Capillary networks connecting the arterioles and venules of the DVP provide nourishment for the adnexal structures within the reticular dermis. The venules of the DVP serve as portals of entry for leukocytes associated with inflammation of the adnexa or for inflammation within the reticular dermis itself, as occurs in erysipelas.
Dermal microvessels are surrounded by flat adventitial cells, ultrastructurally most closely resembling a fibroblast, called veil cells.1 Veil cells are external to the wall, demarcating the vessel from the surrounding dermis. These cells lack leukocyte markers and are negative for HLA-DR and CD1a expression. They express glycoprotein Ib α, von Willebrand factor (vWF) receptor, and factor XIIIa, a coagulation transglutaminase, and are sometimes called XIIIa-positive dendrocytes. Factor XIIIa-positive dendrocyte rarefaction is found in the classic type of Ehlers–Danlos syndrome, but the significance of this finding is not clear. They should be distinguished from dermal dendritic cells, a population of specialized antigen-presenting leukocytes resident within the dermis.
Dermal microvessels, especially capillaries, contain, in addition to ECs, a population of contractile mesenchymal cells called pericytes (PCs). Unlike veil cells, PCs are an integral component of the vessel wall. They extend long cytoplasmic processes over the abluminal surface of the ECs and make interdigitating contacts. The coverage of ECs by PCs varies considerably among different microvessel types. Communicating gap junctions, tight junctions, and adhesion plaques are present at these points of contact.
Because PCs are rich in contractile proteins and have complex interdigitations with ECs in postcapillary venules, it is assumed that they are responsible, at least in part, for gap formation in response to, for example, histamine or bradykinin (although cultured ECs are capable of contraction in the absence of PCs). Moreover, interaction between PCs and ECs is important for the maturation, remodeling, and maintenance of the vascular system via the secretion of growth factors or modulation of the extracellular matrix. There is also evidence that PCs are involved in the transport across the blood–brain barrier and the regulation of vascular permeability.
Morphology and Molecular Markers
ECs usually appear as flattened epithelium-like cells, with a thickness typically less than 10 μm and a surface area covering up to 1,000 μm2 (see Fig. 162-1). In large vessels, ECs have an elongated shape, especially in regions of laminar flow, and have their long axis oriented in the direction of blood flow. ECs located in regions of disturbed flow (i.e., at branch points of the arterial tree) are often polygonal rather than elongated. Cultured ECs are typically polygonal but elongate when flowing medium is passed over them for periods of 24 hours or more.4 Shear stress, the force imparted by viscous drag of flowing liquid, is responsible for inducing a cytoskeletal rearrangement and the change of shape from polygonal to elongated. Interestingly, the microtubular organizing center of ECs, located near the nucleus, is always oriented toward the heart (i.e., upstream of the nucleus in arteries and downstream of the nucleus in veins) regardless of the direction of blood flow.5 Moreover, ECs have an apical/luminal polarization, integrin receptors for matrix macromolecules are concentrated on the basal/abluminal surface, and adhesion molecules for leukocytes are almost exclusively displayed on their apical surface, which faces the lumen of the blood vessel.6
The ultrastructure of a postcapillary venule is shown in Fig. 162-3. ECs have a number of characteristic organelles. The best described are Weibel–Palade bodies (WPBs), fenestrae, and the caveolar system. WPBs are elongated, membrane-bound organelles that display longitudinal striations by electron microscopy. WPBs are distributed randomly throughout the cytoplasm in vascular ECs of most vertebrates. WPBs constitutively contain vWF (also called factor VIII-related antigen), P-selectin (CD62P),7 lysosomal membrane-associated glycoprotein 3 (or CD63),8 1,3-fucosyltransferase VI,9 interleukin 8 (IL-8),10 and angiopoietin 2 (Ang-2).11 The longitudinal striations within the WPBs are formed by highly polymerized forms of vWF, reaching sizes of up to 20,000 kDa. On release, vWF polymers may attach to the basement membrane and stabilize platelet adhesion to the subendothelial matrix, particularly in high-velocity arterial flow. Although vWF is commonly used as an EC marker (see Section “Molecular Markers of Endothelial Cells”), it is also found in megakaryocytes, which appear to lack a physiologic mechanism of release, synthesizing vWF only for storage within the α granules of their platelet progeny. P-selectin is rapidly translocated to the cell surface as the WPB fuses with the plasma membrane, where it mediates leukocyte adhesion to ECs (see Section “Leukocyte Adhesion”). CD63 is a 53-kDa lysosomal membrane glycoprotein belonging to the tetraspanin superfamily; its function in the WPB is unknown.
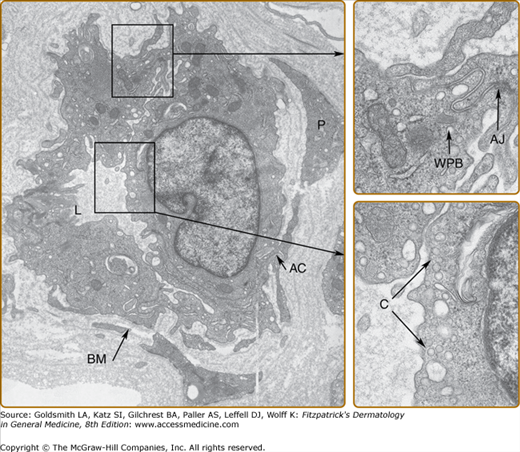
Blood vascular ECs usually form a continuous monolayer, and most transendothelial transport takes place in the junctions between cells. One important exception is in the fenestrae of capillaries in lymph nodes, capillaries in the intestinal mucosa, renal glomerular ECs, and hepatic and bone marrow sinusoids. Fenestrae, uniquely found in ECs, are specialized annular patches of attenuated cytoplasm and membrane, about 175 nm in diameter, characterized by an increased concentration of anionic lipids.12 They group into structures resembling sieve plates that occupy 6% to 8% of the surface capillaries. Fenestrated capillaries are much more permeable to water and small-molecular-size solutes than those of the continuous type. The anionic charge probably precludes the transfenestral passage of anionic plasma proteins. Various factors, such as pressure, alcohol, serotonin, nicotine, and infection, change the number and diameter of fenestrae, and ECs of the continuous type may become transiently fenestrated.
All ECs appear to contain numerous vesicles subjacent to the plasmalemma, both luminal and abluminal. On careful examination, most of these vesicles turned out to have narrow tubular connections to the plasma membrane. Such flask-shaped invaginations are called caveolae, literally “little caves,” and are part of the surface membrane, either as a single caveola or as more complex racemose invaginations. In situ, they may account for 50% or more of the plasma membrane surface area of ECs. Caveolae arise from a late Golgi compartment and represent a stable membrane unit built up around caveolins, cholesterol, and glycosphingolipids. ECs are rich in caveolins 1 and 2. Caveolins bind to cholesterol and are required for caveolae formation; ECs in caveolin 1 knockout mice fail to form caveolae. Expression of caveolin 1 in some cell types normally lacking caveolae can cause their formation, but cultured ECs normally retain expression of caveolins yet lose their caveolae, with cholesterol and glycosphingolipids dispersing into submicroscopic lipid rafts on the now simplified plasma membrane, implying a key role for an as yet unidentified factor.
Caveolae can mediate vesicular transport, internalize certain plasma membrane proteins and provide a platform for the assembly of signaling complexes at the surface of the cell. Caveolins interact with a variety of downstream signaling molecules, including Src-family of tyrosine kinases, p42/44 mitogen-activated protein kinase, and endothelial NO synthase (eNOS). They hold these signal transducers in the inactive conformation until activation by an appropriate stimulus. For example, the amount of caveolin is inversely related to NO production by eNOS.13 Mice lacking caveolin 1 have impaired NO and calcium signaling in the cardiovascular system, which causes aberrations in EC-dependent relaxation, contractility, and maintenance of myogenic tone.14
Caveolin 1 is associated with a number of growth factor receptors, including the epidermal growth factor receptor, the high affinity nerve growth factor receptor (TrkA), the insulin receptor, the platelet-derived growth factor receptor and transforming growth factor receptors I and II.15,16 In addition, cytokine receptors like the tumor necrosis factor (TNF) receptor 1 (CD120a) and interferon receptors also localize to caveolae. For these receptors, the invaginated structure of caveolae provides an optimal environment for receptor function, forming a more stable signaling complex and allowing for cross talk with other receptors.
The lipid constituents of caveolae are the same as those of lipid rafts, but rafts are regions without membrane indentations and they are flat. A key limitation in a clear distinction between rafts and caveolae is due to methodical limitations to clearly separate them. Moreover, rafts and caveolae should be considered as dynamic entities that form and dissipate in response to various stimuli, and both may facilitate transport of entities to other cellular regions and across the cell.
Another organelle that is present in venular endothelium is so called vesicular–vacuolar organelle, grapelike structures of interconnected vesicles that extend throughout the cytoplasm. They show a wide variation in size and most vesicles do not contain caveolin. Consistent with this finding, they are present in normal size and numbers in caveolin null mice.17 These organelles appear to be critically involved in acute vascular hyperpermeability, which typically involves the postcapillary venules, where these vesicular–vacuolar organelles are typically situated.
Neighboring ECs connect to each other by forming interdigitating protrusions connected by junctions. Junctions possess different degrees of complexity along the vascular tree, reflecting different permeability for proteins and cells. Intercellular tight (occluding) junctions constitute the anatomic basis for the tightly regulated interfaces of the blood–testis and blood–brain barriers.18 They consist of a continuous belt-like meshwork of six anastomosing junctional strands, which prevent the transport of macromolecules into the tissue. Arterial ECs also possess tight junctions19; however, they consist of only one or two junctional strands, and tight junctions are difficult to find in postcapillary venules. Tight junctions contain a complex of transmembrane (junctional adhesion molecule-1, occludin, and claudins) and cytoplasmic (zonula occludens-1 and -2, cingulin, AF-6, and 7H6) proteins linked to the actin cytoskeleton. The claudin multigene family encodes tetraspan membrane proteins that are crucial structural and functional components of tight junctions. In mammals, the claudin family consists of 24 members, which exhibit complex tissue-specific patterns of expression. Their extracellular loops interact with each other to seal the cellular sheet and regulate paracellular transport between the luminal and basolateral spaces. The transmembrane protein occludin is linked to ZO-1, a cytoplasmic protein that interacts with the cytoskeleton and appears involved in opening and closing of the junctions. Junctional adhesion molecules, called JAMs, members of the immunoglobulin superfamily, form homophilic adhesion sites in tight junctions and play a role in the generation of cell polarity, keeping luminal proteins from diffusing to the abluminal cell surface and vice versa.
Adherens junctions are found in all vascular ECs (see Fig. 162-3). They are multimeric protein structures that mediate homotypic cell adhesion. In endothelial cells, they are distributed along the cleft between neighboring cells and are frequently intermingled with tight junctions. During embryonic development, this type of junction promotes adhesion of identical cells and is important for organization and separation of tissues. In the adult, they maintain tissue homeostasis and contribute to paracellular permeability. Moreover, they confer cell-to-cell signals such as contact inhibition of cell growth, or resistance to apoptosis. They also regulate cell shape and polarity. Endothelial adherens junctions are principally organized by VE-cadherin [also called cadherin 5 or vascular endothelial cadherin (CD 144)]. VE-cadherin-negative ECs derived by gene targeting fail to organize in vessel-like structures, blocking VE-cadherin interactions with monoclonal antibodies inhibits vessel formation.20,21 Whereas VE-cadherin appears critical for initial formation of blood vessels, for maintenance and remodeling of blood vessels, a receptor-type phosphotyrosine phosphatase (RPTP) called VE-PTP appears essential. VE-PTP binds to VE-cadherin via the extracellular domain and supports adhesive functions of VE-cadherin.22 Intracellularly, VE-cadherin is linked to a large cytosolic protein complex. This complex contains catenins, β-catenin, p120ctn, plakoglobin (also called γ-catenin), and α-catenin. Catenins β-catenin, p120ctn and plakoglobin contain homologous Armadillo repeats, whereas α-catenin is homologous to vinculin, which is another actin-binding protein. Moreover, this complex contains src, fyn and src homology-containing phosphatase (SHP)-1, and RPTP-μ and may associate with neighboring transmembrane receptors such as vascular endothelial growth factor receptor (VEGF-R)-2, angiopoietin receptor 1 (also known as Tie-2) and protease activated receptors (PAR)-3 and -6. Thus, adherens junctions integrate signals from neighboring cells and neighboring transmembrane receptor molecules and link these signals to the cytoskeleton and through focal adhesion kinase with integrin receptors to the basal matrix. This signaling pathway also functions retrograde from integrin receptors to adherens junctions. This network gets further input through RhoGTPases signaling to Rho kinase (ROCK) and leading to contraction of the cytoskeleton.17,23–29
ECs form gap junctions, demonstrated by transfer of microinjected small-molecular-weight tracers between adjacent ECs. In ECs, the relevant gap junctional proteins are connexin-43, connexin-37, and connexin-40. Interestingly, ECs also may form heterogap junctions with PCs or SMCs in the vessel wall and sometimes with adherent leukocytes.
ECs rest on a basal lamina, which is formed by EC itself and by surrounding cells. It consists of laminins, collagens, fibronectin, nidogen (entactin), and heparan sulfate proteoglycans. Interaction with these matrix molecules is essential for maintaining normal endothelial function. For example, the laminin A chain peptide supports endothelial branching and formation of new capillaries.30 Furthermore, a 20-kDa C-terminal fragment of collagen XVIII specifically inhibits endothelial proliferation and angiogenesis,31 which suggests that collagen XVIII is also a positive regulator of vessel growth. On the other hand, EC–collagen V interactions inhibit endothelial attachment and growth. ECs interact with these components of the basal lamina by numerous transmembrane adhesion molecules. The complexities of these interactions have only been partially characterized. Members of the integrin family are particularly important for this function. For example, α6β1-, α2β1-, α5β1-, and αVβ3-receptor complexes are important for endothelial attachment to, and migration on, laminin, collagen, and tenascin, and therefore serve important roles in angiogenesis. In addition, integrin binding to matrix proteins also has a multitude of intracellular effects on the organization of the endothelial actin skeleton and on signaling processes within the cell.3 Endothelial-cell growth factors have been shown to differentially upregulate the biosynthesis of α2, α5, β1, and β3 integrins, serving the different needs of endothelium during growth, spreading, and new vessel formation. Moreover, integrins show certain tissue-and vessel-restricted expression; for example, α1β1 is found only in small blood vessels and capillaries but not in large vessel ECs. Matrix proteins such as thrombospondin 1, may also serve as inhibitors of angiogenesis.33
ECs display a number of molecular markers that result in a characteristic profile detectable with antibodies, lectins, and other ligands (Table 162-1). In addition to containing molecules expressed on all endothelium, the skin vasculature shows several site-specific characteristics. Postcapillary venules of the SVP are CD36− and thus distinct from capillaries of the DVP and from capillaries of other tissues, which are CD36+.34 CD36 antigen is a cellular receptor for thrombospondin, collagen, low-density lipoproteins, long-chain fatty acids, and platelet-agglutinating protein p37.35 Moreover, CD36 functions as one of the receptors that mediate the adhesion of Plasmodium falciparum-infected erythrocytes to microvascular EC. The functional consequences of the disparate CD36 expression on SVP and DVP endothelium are unknown. Microvessels are best characterized by expression of the pathologische anatomie Leiden-endothelium (PAL-E) antigen (which is identical with plasmalemmal vesicle 1 or fenestrated endothelial-linked structure protein),36 N-cadherin, and CXC chemokine receptor 4 (CXCR4), and by their surrounding by smooth muscle actin-positive cells (which may be SMCs or PCs). Blood vascular ECs normally lack the lymphatic EC markers lymphatic vessel endothelial-1 [LYVE-1, (a hyaluronan receptor) and podoplanin], but may express these molecules when inflamed.37,38 ECs express receptors for binding members of the VEGF family. Blood endothelial cells express VEGFR-2 and VEGFR-1, whereas lymphatic vessel endothelial cells express VEGFR-2 and VEGFR-3.39–42 Neuropilins, another group of VEGF-binding membrane-bound receptors, are also differentially expressed in various types of vasculature. Whereas neuropilin-1 is strongly expressed in arterial endothelial cells, neuropilin-2 is expressed in veins and visceral lymphatic vessels.43 Another important signaling system is the angiopoietin/Tie receptor system (see Section “Regulation of Angiogenesis and Vasculogenesis”).
Marker | Location/Function | Expression Profile |
|---|---|---|
VWF | WPB, biosynthetic organelles (Golgi, ER) | EC, megakaryocytes, platelets |
CD31 | Adhesion molecule, transmigration | EC, platelets, leukocytes |
CD143 (angiotensin-converting enzyme) | Secreted then bound to cell surface | EC, macrophages |
CD141 | Anticoagulant thrombomodulin | EC, trophoblast of the placenta |
Reactivity with the lectin UEA-1 | Blood group H antigens | EC, erythrocytes, specialized epithelia |
VEGFR-1 and VEGFR-2 | Receptor tyrosine kinase | EC, macrophages, hematopoietic stem cells, motor neurons (R2) |
Tie-1 and Tie-2 | Receptor tyrosine kinase | EC, hematopoietic stem cells, peripheral nervous tissue (Tie-2) |
ICAM-2 | Adhesion molecule | EC |
CD40, CD54, CD58, CD105 | – | EC, broad expression pattern |
CD9, CD13, CD31, C1q and C5a receptors, complement regulatory proteins | Common embryologic origin of the angioblast and the hematopoietic stem cell | EC, hematopoietic cells |
CD26, CD40, CD44, CD47, CD54, CD58, CD146 | — | EC, broadly expressed on other cell types |
Class II MHC molecules | Antigen presentation | Constitutive on skin and kidney microvessels |
Endoglin (CD105) | Surface receptor | EC, stromal cells |
PAL-E antigen | Fenestrae | Microvessels and veins |
CD34 | Proteoglycan, adhesion molecule | Microvessels, hematopoietic cells |
CD36 | Proteoglycan | Microvessels except brain and some skin ECs |
CD32 | Fc receptor | Liver and skin microvessels, leukocytes |
P-selectin | Prestored in WPB | EC |
VCAM-1 (CD106) | Inducible adhesion molecule | EC, macrophages, follicular-dendritic cells |
E-selectin (CD62E) | Inducible adhesion protein | EC |
MAdCAM-1 | Adhesion molecule, sialomucin | Microvessels of intestine and breast |
GlyCAM-1 | Adhesion molecule, sialomucin | Lymph nodes, high endothelial venules |
| Peripheral node addressing (antibody MECA-79) | L-selectin ligands | EC of peripheral nodes and skin |
ICAM-3 | Adhesion molecule | Tumor vessels |
Postcapillary venules of the skin express toll-like receptor (TLR) family members and CD32 molecules (FcγRIIa).44 CD32 binds complexed immunoglobulin G and is thought to function in the elicitation of type III (immune complex-mediated) hypersensitivity as well as in immune complex clearing. ECs of postcapillary venules express significantly higher levels of the histamine H1 receptor than other ECs. Histamine produces increased albumin leakage and induces rolling of leukocytes, mediated in part by P-selectin expressed on the EC surface. PAR 2 expression at this site also plays a role in inflammatory reactions. PAR 2-activating peptides increase leukocyte rolling and adhesion to postcapillary venules. Finally, postcapillary vessels of the skin in situ constitutively express major histocompatibility complex (MHC) class I and II molecules at their luminal surfaces, which implies that ECs may directly participate in the elicitation of antigen-driven immune responses. As discussed later (see Section “Role of Endothelium in Adaptive Immunity”), skin microvessels in cell culture appear able to process and present antigen in an MHC-restricted manner to T cells.45
Endothelium in Inflammation
Innate immunity, also called natural or native immunity, comprises the host’s defense systems against microbes that are independent of T and B lymphocytes and their receptors for specific antigens (see Chapter 10). The innate immune system is much older in evolutionary terms than the T- and B-cell-specific immune system (often called adaptive immunity to distinguish it from innate immunity). The humoral component primarily involves the complement system. The primary cellular effectors of innate immunity in humans are neutrophils, mononuclear phagocytes, and natural killer cells, but ECs also participate.
Endothelial cells represent the initial target in many different types of infection.46 For example, exposure to lipopolysaccharide, a major cell-wall constituent of Gram-negative bacteria, results in endothelial activation through a receptor complex consisting of TLR-4, CD14, and MD2. Then, recruitment of the adaptor protein myeloid differentiation factor (MyD) 88 to the ligand-activated TLR-4 receptor initiates an MyD88-dependent pathway that culminates in the early activation of nuclear factor-κ B and the mitogen-activated protein kinases. In parallel, an MyD88-independent pathway using an alternative adaptor protein called TRIF results in a late-phase activation of nuclear factor-κ B as well as generation of interferons.47,48 ECs also express TLR-2, which serves as a signaling receptor for Gram-positive cell-wall components, and intracellular TLRs that respond to various types of nucleic acids typically encountered during infections by viruses. Stimulation of TLR 2/6 by bacterial lipopeptides also may promote angiogenesis and results in the secretion of GM-CSF.49 Certain vascular beds express the macrophage mannose receptor (CD206), a receptor for oligosaccharides on the cell wall of bacteria, yeasts, and parasites. Other receptors of the innate immune system include a variety of lectin-type molecules, such as mannose-binding protein or galectins that recognize bacterial carbohydrates. Mannose-binding protein may stimulate the alternative pathway of complement activation.
TNF and IL-1 are the principal effector cytokines of the innate immune response to bacteria. In general, endothelial responses to TNF and IL-1 are similar and are considered together. The major isoform of IL-1 that is produced during innate immune responses is IL-1β, but IL-1α, the major isoform made by ECs, has indistinguishable actions, and we will simply refer to both isoforms as IL-1. At least five changes in the phenotype of ECs that promote inflammation, collectively called endothelial activation, have been described.
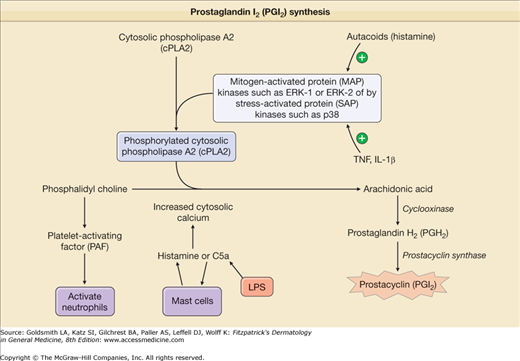
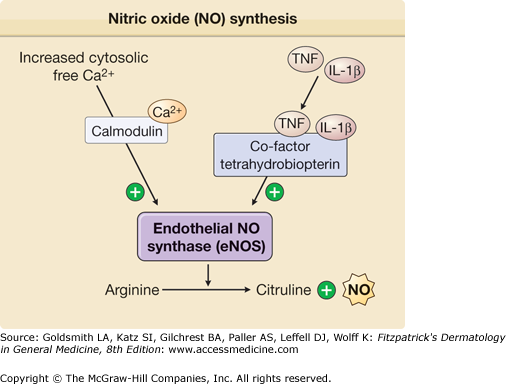
TNF- or IL-1-treated ECs increase production of vasodilators, such as PGI2 and NO, that relax smooth muscle cell tension, thereby producing vasodilation and increasing tissue perfusion. PGI2, also called prostacyclin, is synthesized in ECs by a series of three sequential enzyme-catalyzed reactions.52 ECs express one isoform of prostaglandin H synthase, PGHS-1, also called cyclooxygenase 1, that is constitutively active. Treatment of ECs with TNF or IL-1 dramatically increases PGHS activity by inducing de novo expression of a second isoform, PGHS-2, which is also called cyclooxygenase 2. This response markedly increases the capacity to make prostaglandin (PG)H2 from arachidonic acid, and the increased PGH2 is subsequently converted into PGI2 (Fig. 162-4). Pharmacologic inhibition of the synthesis of PGH2 is thought to contribute to the anti-inflammatory actions of aspirin and nonsteroidal anti-inflammatory drugs. NO is produced by conversion of arginine to citrulline through eNOS (Fig. 162-5). TNF exhibits opposing effects on vasodilation. Initially, it enhances eNOS function and NO synthesis through activation of Akt and by increasing the synthesis of tetrahydrobiopterin, a limiting cofactor for eNOS function.53 At later times, TNF destabilizes eNOS mRNA and thereby inhibits NO production.
(Tables 162-2 and 162-3). TNF- or IL-1-treated ECs exhibit changes in the cell surface that favor the initial capture, rolling, and, ultimately, firm adhesion and spreading of leukocytes (Fig. 162-6). Adhesion is a necessary prelude to extravasation of leukocytes into the tissues. Capture and rolling are typically mediated by selectins expressed on the leukocyte or on the EC.54 There are three structurally related selectin proteins: (1) L-selectin (CD62L), (2) E-selectin (CD62E), and (3) P-selectin (CD62P). In the presence of flowing blood, the leukocyte is propelled in the direction of blood flow, rapidly breaking and reforming selectin-mediated attachments. This results in rolling of the leukocyte along the EC surface (see Fig. 162-6). Ligands involved in selectin-mediated capture may not be the same as those involved in selectin-mediated rolling; for example, neutrophils may switch from L-selectin to E-selectin ligand (ESL)-1 as the principal E-selectin ligand during these two phases of the interaction.55,56 Studies using blocking monoclonal antibodies or selectin knockout mice have confirmed that selectins are required for neutrophil rolling and subsequent recruitment in vivo. In mice, E- and P-selectin functions may be redundant (i.e., both molecules must be knocked out or inhibited for complete loss of rolling). Of note, whereas IL-1 and TNF upregulate both P- and E-selectin in mice, they induce only E- but not P-selectin in humans. In humans, E-selectin expression is largely regulated by de novo synthesis induced by cytokines whereas P-selectin is preformed and mobilized from WPBs to the cell surface in response to stimuli like thrombin, histamine, reactive oxygen species, and the like. Consequently, E- and P-selectin may not be redundant in humans. Selectin expression is rapid, within 5 min for P-selectin and 1 to 2 hours for E-selectin, but is often transient, peaking at 15 minutes for P-selectin and 4 to 6 hours for E-selectin, but may persist much longer on skin ECs. Another important mechanism initiating rolling of T lymphocytes on ECs is the interaction of the hyaluronan receptor CD44. CD44 is expressed on endothelial cells and, under inflammatory conditions, binds hyaluronan, which then binds CD44 expressed on activated T lymphocytes and macrophages.57
Adhesion Molecule | Expression | Ligand | Function |
|---|---|---|---|
Selectins | |||
L-selectin (CD62L) | Leukocytes | Sialylated and fucosylated carbohydrates displayed on, for example, CD34, MAdCAM-1, or GlyCAM-1 | Tether, rolling |
E-selectin (CD62E) | EC | Sialylated Lewis X or A moieties such as CLA displayed on ESL-1, L-selectin or PSGL-158 | Tether, rolling |
P-selectin (CD62P) | EC, platelets | Glycans and sulfated sialomucins displayed onPSGL-195 | Tether, rolling |
Integrins | |||
LFA-1 (αLβ2 or CD11a/CD18) | Leukocytes | ICAM-1, ICAM-296 | Firm adhesion, diapedesis |
Mac-1 (αMβ2 or CD11b/CD18) |








