Fig. 23.1
Keratinocyte culture IC50 and estimated unbound epidermal concentrations after human epidermal membrane penetration of sunscreens from a 1 % mineral oil solution (Adapted from Hayden et al. (2005))
These findings can be summarized by defining a cutaneous margin of safety after topical application as the ratio of concentration of active and excipient associated with toxicity (e.g. IC50) to the viable epidermal concentration of the active or excipient. A desirable margin of safety based on human models should be at least 10 to account for inter-subject variability. Toxicities, such as irritancy, photosensitization and corrosivity, are likely to be better defined by the probability of an event occurring in a given patient after a particular exposure and are dependent on patient susceptibility and the nature and conditions of the product used.
The second safety measure of interest is the systemic margin of safety which arises when an active applied to the skin for local cutaneous effect enters the systemic circulation and causes systemic side effects. This concept is most highly developed for topically applied corticosteroids and, in this case, is quantified by the extent of suppression of the adrenal gland in the hypothalamic–pituitary–adrenal axis. A systematic literature review of the risk of adrenal axis suppression and skin atrophy after application of topical corticosteroids in plaque psoriasis for the period 1980 to January 2011 found morning cortisol was reduced in 0–48 % of patients in 11 short-term studies (Castela et al. 2012). Thong et al. (2007) have listed a range of topical drugs and chemicals known to cause systemic effects. The agents include anti-infectives, antihistamines, minoxidil, insecticides, solvents and steroids.
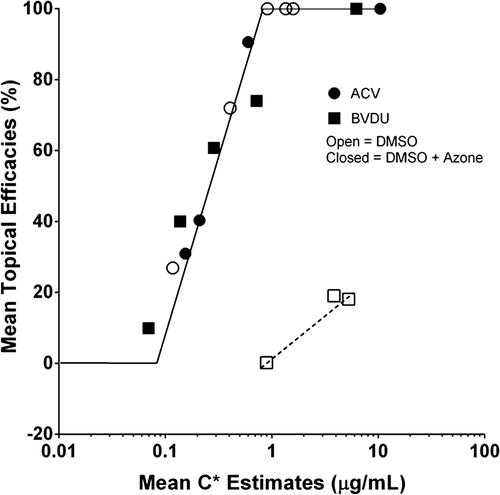
The corollary measure to safety is cutaneous efficacy and, in general, the more potent the topical product used, the less skin flux required to achieve a desired local cutaneous effect after topical application. A commonly used approach, analogous to that described earlier for toxicity, is to estimate the free drug concentration at the local target site (C *), which, for antivirals, is assumed to be the epidermal basal cell layer (Patel et al. 1996). Afouna et al. (1998) analysed the receptor solutions for full thickness hairless mouse skin mounted in Franz diffusion cells. Assuming that receptor sink conditions applied, C * was estimated as the ratio of the steady state penetration flux (J s) to in vivo dermis permeability coefficient (k p, d ):
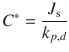
These C* values derived for various formulations of bromovinyldeoxyuridine (BVDU) and acyclovir (ACV) were then compared with the in vivo efficacy of the formulations against cutaneous herpes simplex virus type 1 infections in hairless mice. Figure 23.2 shows a composite graph of their findings. It is evident that a similar concentration response relationship exists for the 95 % dimethyl sulfoxide (DMSO) + 5 % Azone for both BDVU and ACV and for the 95 % DMSO only formulation for ACV, whereas much higher BVDU C* values are needed for the BVDU in 95 % DMSO only formulation. They surmise that Azone could have two effects. Firstly, it may either enhance penetration of the active. This is evident from the 6.5 times higher C* for 5 % ACV in 95 % DMSO +5 % Azone than in the 95 % DMSO only formulation. The effect is less for BVDU, where the C* for 5 % BVDU in 95 % DMSO +5 % Azone is 1.65 times that of 95 % DMSO only formulation. Secondly, Azone may have a synergistic effect with the antiviral agents on viral kill, evident in the shift towards higher potencies of C* for BVDU but not for ACV (Fig. 23.2).
(23.1)
Fig. 23.2
Correlation of estimated values of C* from in vivo–in vitro experiments with observed in vivo efficacies for 9 acyclovir (ACV) formulations and 8 bromovinyldeoxyuridine (BVDU) formulations. Open symbols: formulations in DMSO without azone; closed symbols: formulations in DMSO with 5 % azone (Adapted from Afouna et al. (1998))
A key assumption made in this analysis is that the dermal permeability coefficient (which is normally expressed in the pharmacokinetic literature as a dermal clearance (Cld) (Siddiqui et al. 1989) (i.e. 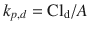 , where A is the surface area of application) can be estimated by the ACV skin penetration flux that yields 50 % topical efficacy (J 50 ) divided by the free plasma levels of ACV that inhibit 50 % of cutaneous lesions treated systemically with ACV (C * 50), i.e.
, where A is the surface area of application) can be estimated by the ACV skin penetration flux that yields 50 % topical efficacy (J 50 ) divided by the free plasma levels of ACV that inhibit 50 % of cutaneous lesions treated systemically with ACV (C * 50), i.e. 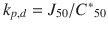 . It is also evident that systemic efficacy of various formulations is much less than when applied topically, showing the enhanced response that can be achieved by targeted topical application.
. It is also evident that systemic efficacy of various formulations is much less than when applied topically, showing the enhanced response that can be achieved by targeted topical application.
, where A is the surface area of application) can be estimated by the ACV skin penetration flux that yields 50 % topical efficacy (J 50 ) divided by the free plasma levels of ACV that inhibit 50 % of cutaneous lesions treated systemically with ACV (C * 50), i.e. . It is also evident that systemic efficacy of various formulations is much less than when applied topically, showing the enhanced response that can be achieved by targeted topical application.Davis has recast the free drug concentration approach described above in Eq. (23.2), which describes the efficacy of the active in terms of both potency of the active and how much penetrates to a target site (Davis 2008; Jepps et al. 2013):

A major issue not fully recognized in the work of Cordero and Davis (Cordero et al. 2001; Davis 2008) is that the fraction of active bound in the in vitro system used to determine potency may differ from that present in skin penetration studies and in vivo. In order to simplify the present description and be consistent with the work reported by Cordero and Davis, this important correction will not be included in the following discussion. Hence, if a cutaneous concentration for a desired effect is defined as C eff and that observed after topical application as C ve, the efficacy is given by the ratio C ve/C eff. C ve is defined by the penetration flux divided by the clearance from the viable epidermis. Cordero et al. assumed that this clearance, in turn, was related to the in vitro dermis diffusion and thickness. In reality, as discussed earlier in referring to the work of Afouna et al (1998), this should be in vivo clearance. Our own work supports this, suggesting that, firstly, uptake by the blood in the dermal capillaries located just below the viable epidermis is likely to be the rate limiting determinant of clearance in vivo (Cross and Roberts 2006) and secondly, carriage of topical non-steroidal anti-inflammatory drugs (NSAIDS) to deeper tissues below that application site is also dependent on blood flow for highly plasma protein-bound drugs (Dancik et al. 2013a, b). Accordingly, we have, for illustrative purposes, assumed both constant binding and clearance and expressed efficacy simply as a ratio of flux divided by the in vitro minimum inhibitory concentration (MIC), as originally proposed by Mertin and Lippold (Mertin and Lippold 1997).
(23.2)
Thus, a greater efficacy is provided by an active with either a higher flux or a greater potency (a lower dose for the same effect). These concepts are illustrated in Fig. 23.3, where data from Cordero et al. and others for saturated skin flux (Fig. 23.3a), anti-inflammatory activity (Fig. 23.3b) and efficacy (Fig. 23.3c) for a range of non-steroidal anti-inflammatory drugs are shown (Mertin and Lippold 1997; Wenkers and Lippold 2000; Cordero et al. 2001; Jepps et al. 2013).
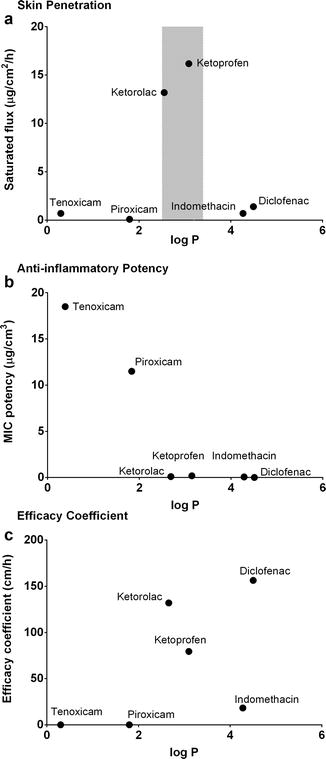
Fig. 23.3
(a) Saturated skin flux, (b) in vitro anti-inflammatory activity (MIC) and (c) likely clinical efficacy of various non-steroidal anti-inflammatory drugs, expressed as an efficacy coefficient, calculated as saturated flux/MIC potency for a range of non-steroidal anti-inflammatory drugs with varying values for logarithm octanol–water partition coefficient (log P). Flux and MIC data from Cordero et al. (2001) (closed symbols) (Adapted from Jepps et al. (2013))
In Fig. 23.3a, the shaded area indicates the region of optimal skin penetration occurring at a log P of around 3 (Yano et al. 1986; Zhang et al. 2009). Ketorolac and ketoprofen, in this range, consequently have the greatest saturated flux. The more lipophilic actives also have a greater in vitro performance index of anti-inflammatory activity (i.e. lower MIC), and so these two compounds also have high efficacy coefficients. Diclofenac, although having a less favourable log P of >4, still has the highest predicted efficacy coefficient as a consequence of having the lowest MIC. On the other hand, piroxicam has the least saturated flux of all these actives, as well as a relatively high MIC, and consequently has the lowest efficacy coefficient of these non-steroidal anti-inflammatory drugs (NSAIDS). Apart from a comparatively higher molecular weight and melting point, hydrogen bonding seems to be playing a pivotal role in the lower flux of piroxicam (Anrade and Costa 1999).
Adrian Davis (Davis 2007) also presented efficacy and systemic toxicity indices for topical immunosuppressive agents used in treating atopic dermatitis, extracted from Trottet’s PhD thesis (Trottet 2004) at the SCI meeting in 2007 (see Fig. 23.4). The corresponding margins of safety for cyclosporin A, tacrolimus and pimecrolimus, calculated as the ratio of the systemic safety indices and the efficacy indices, were 2400, 47 and 457, respectively.
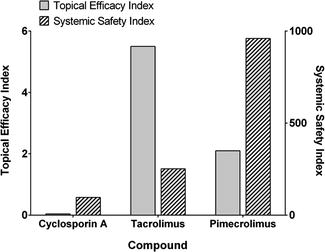
Fig. 23.4
Comparison of topical efficacy and systemic safety indices for topical immunosuppressive agents, cyclosporin A, tacrolimus and pimecrolimus treatment in atopic dermatitis. The corresponding margins of safety are 2400, 46 and 457, respectively (Adapted from Davis (2008))
23.1.2 Systemic Concentration of Solutes and Their Effect on Active Performance
The systemic efficacy of many actives, such as anticonvulsants, anti-infective and cardiac drugs, can be defined by plasma concentrations after dosing by a particular route of administration. As shown in Fig. 23.5, in transdermal delivery occurring from a constant rate transdermal passive delivery patch, there is a lag prior to reaching maximal levels and in returning to baseline on patch removal (Roberts and Walters 2007). Further, after reaching a maximum, the serum levels slowly decline with time, consistent with a reduction in flux due to a gradual depletion in the amount of active in the patch.
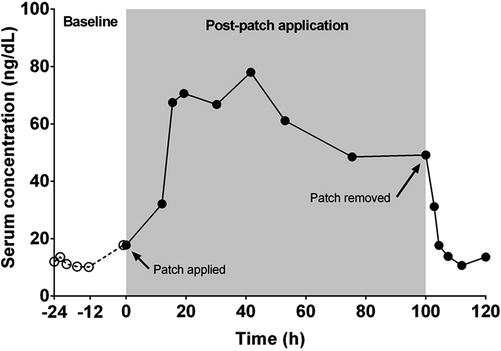
Fig. 23.5
Mean total testosterone concentrations in serum over time, following application of a single 28 cm2 patch for 96 h, systemically delivering testosterone at a rate of approximately 300 μg/day (Adapted from Roberts and Walters (2008))
Topical products, especially transdermal patches, are often required to provide constant therapeutically effective plasma concentrations, C ss. The penetration flux J s ideally needed to reach such concentrations is given by Eq. (23.3), where Clbody is the body clearance and F s is the availability
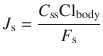
Table 23.1 shows some estimated transdermal fluxes required for the topical administration of a number of pharmaceuticals used in transdermal systems, by substituting desired plasma concentrations and body clearances into Eq. (23.3).
(23.3)
Table 23.1
Transdermal delivery
Solute | Indication | Molecular weight | Melting point (°C) | log K oct | t ½ | Plasma level (μg/h) | F s | CLbody L/h/70 kg | Estimated J s,a required (μg/h) |
|---|---|---|---|---|---|---|---|---|---|
Buprenorphine | Pain relief | 468 | 209 | 3.44 | 4 | 0.1–0.52 | 0.5 | 76 | 15–80 |
Clonidine | Hypertension | 230 | 140 | 1.77 | 6–20 | 0.2–2.0 | 1.0 | 13 | 2.7–28 |
Estradiol | Female hormone replacement | 272 | 176 | 2.69 | 1 | 0.04–0.15 | 0.16 | 4–7 (CL/F) | 1–6 |
Ethinyl estradiol | Female contraception | 296 | 143 | 4.52 | 17 | 0.011–0.137 | 1 | 28 | 0.31–3.8 |
Fentanyl | Chronic pain | 337 | 83 | 4.37 | 17 | 1 | 1 | 25–75 | 25–75 |
Isosorbide dinitrate | Angina | 236 | 68 | 1.31 | 105 | 22 | 1 | 1.22 | 28 |
Methyl phenidate | ADHD | 233 | 74.5 | 2.55 | 3–5 | 20–46 | 1 | 60 L/h/30 kg in children | 720–2160 |
Nicotine | Smoking cessation | 162 | ~80 | 1.17 | 2 | 10–30 | 1 | 72 | 900–2630 |
Nitroglycerin | Angina | 227 | 13.5 | 1.62 | 0.04 | 1.2–11 | 1 | 13.5 | 16.2–148.5 |
Norelgestromin | Contraception | 327 | 131 | 4.40 | 28 | 56–420 | |||
Norethindrone acetate | Contraception | 340 | 162 | 3.99 | 9 | 2–15 | 1 | 28 (CL/F = 25 L/h) | |
Oxybutynin | Enuresis | 357 | 130 | 5.19 | 2 | 3–4 | 1 | 25–34 | 75–135 |
Rivastigmine | Alzheimer’s | 250 | 124 | 2.14 | 1–4 | 2–10 | 1 | 130 | 260–1300 |
Rotigotine | Parkinson’s disease | 315 | 141 | 4.97 | 5–7 | 0.2–0.6 | 1 | 630 | 126–378 |
Scopolamine | Motion sickness | 303 | 59 | 1.23 | 1.2–2.9 | 0.04 | 1 | 67–205 | 2.6–8.1 |
Selegiline | Depression | 187 | 138 | 2.95 | 2 | 2 | 1 | ||
Testosterone | Hypogonadism | 288 | 153 | 3.31 | 2.3 | 60–100 | 1 | 3–5 | 180–500 |
Timolol | Hypertension | 316 | 72 | 2.46 | 4.1 | 5–15 | 0.76 | 38 | 250–750 |
Triprolidine | Antihistamine | 278 | 60 | 4.22 | 2–6 | 5–15 | 1 | 43.7 | 218–655 |
23.1.3 Enhancing Efficacy by Increasing Fractional Solubility of an Active in the Formulation
A key goal in formulating an active in a transdermal product is to obtain a desired steady state skin penetration flux (amount penetrated over a period of time) of the active. J s is defined by Eq. (23.4) (Reeve et al. 2013), where J sat is the flux from a saturated solution (often referred as maximum flux (Magnusson et al. 2004)), C v is concentration of the active in a given formulation and S v is the solubility of the active in the formulation. f v represents the factional solubility of the active in that formulation, 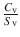 .
.
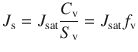
.(23.4)
As is evident from Eq. (23.4), one way to maximize skin penetration is to use a fractional solubility approaching unity. Poulsen led a range of studies that showed maximal skin penetration will occur when an active is at saturation in a formulation (Poulsen et al. 1978).
As discussed in depth by Davis (2008), if an active is used at a concentration below its solubility in the vehicle, the product will normally have a lower efficacy than if the product was used at saturation. Consequently, products may be formulated to contain more active than will actually be needed for an adequate effect, as is the case with transdermal fentanyl patches, which have led to numerous deaths. The danger inherent to fentanyl is its potency (greater than 50–100 times that of morphine) and rapidity of action (Jumbelic 2010). An optimal dosing strategy demands that the rate of active penetration into the skin will achieve and sustain biologically active free levels at the target site within the specified limits. Also, ideally, these conditions should be independent of skin site and skin condition. This is a considerable challenge, given that there is wide variability in skin permeation, depending on the anatomical site (Rougier et al. 1988) and the condition of the skin, such as in particular disease states (Elias and Feingold 1992) or levels of hydration (Roberts and Walker 1993).
As discussed in other chapters in this book, another strategy to increase efficacy is to use a saturated active solution in the vehicle and to further increase either the stratum corneum solubility S sc and/or its diffusivity D sc using co-solvents and enhancers, in accordance with Eq. (23.5), where h sc is the stratum corneum thickness:
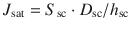
However, care has to be applied in representing such saturated fluxes as maximum fluxes because increasingly, formulations are being designed to include a volatile component that may result in enhanced skin penetration due to the generation of a transient supersaturated state. Many researchers have now promoted the use of supersaturated solutions (Coldman et al. 1969), with several showing that these can lead to even higher fluxes than those achieved with saturated solutions (Lippold 1992; Morgan et al. 1998; Iervolino et al. 2001; Timothy et al. 2002; Santos et al. 2012). More recently, there is an increasing tendency to either stabilize supersaturated products by using polymers (Raghavan et al. 2003) or to generate supersaturation on application to the skin as a consequence of the evaporation of the volatile components in the product (Hadgraft and Lane 2011). Supersaturated systems may be obtained either by design or via solvent evaporation or by mixing of co-solvents. The stability of supersaturated solutions is an important issue, and it would be useful to develop supersaturated systems that are stable throughout the product shelf life. Santos et al. (2011) investigated the permeation of fentanyl from supersaturated formulations across silicone membranes. Supersaturated formulations containing either propylene glycol (PG)/water or PG/ethanol were prepared with varying degrees of saturation (DS) of fentanyl. Both PG/water and PG/ethanol formulations showed good correlation between the flux and DS. The enhancement observed for PG/ethanol formulations confirmed that enhanced active thermodynamic activity was induced due to ethanol evaporation. In further studies, tape-stripping was used to show that supersaturation of the active is maintained in the outer layers.
(23.5)
However, if the active lacks potency, a penetrating active will still prove to be inefficacious.
Obviously, an improved evaluation of product efficacy and safety will contribute to better therapeutic outcomes. Whereas the determination of bioequivalence for two solid oral dosage forms is based on a comparison of active and/or its metabolite concentrations in blood or urine after dosing from a generic product or that of an innovator in healthy volunteers, such considerations are really only appropriate for transdermal products meant for systemic effect. For most topically acting actives, such an approach is not appropriate as the site of action is local, not systemic. Hence, generic dermatological products are only required to demonstrate bioequivalence in clinical trials, including the vasoconstrictor test used to assess the efficacy of topical corticosteroids (Shah et al. 1998). Franz et al. (2009) suggested that a substitute for clinical bioequivalence testing is to define the penetration fluxes of an active through excised human skin. Their evaluation, based on glucocorticosteroids and generic tretinoin gels, indicated that the in vitro model had greater sensitivity than the clinical method in detecting small differences between two products (Franz et al. 2009). It is unclear, however, how this approach can overcome the well-known wide variability in excised human skin permeability. More recently, Lehman and Franz have assessed the bioequivalence of topical retinoid products using pharmacodynamic assays (Lehman and Franz 2012).
23.2 Role of Intended Targets on Topical and Transdermal Delivery
There is a range of skin and systemic sites often targeted by topical and transdermal delivery, summarized in Fig. 23.6. In this section, we illustrate some of the actives and their formulations used to achieve appropriate targeted delivery by topical or transdermal delivery.
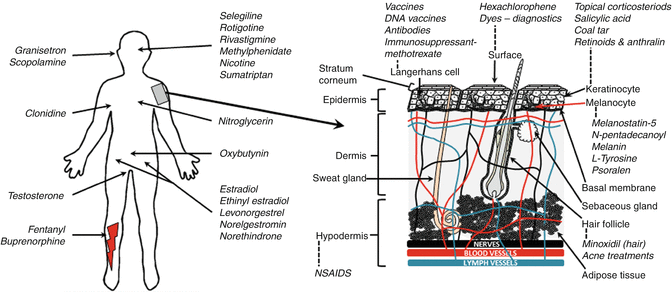
Fig. 23.6
Target organs and sites for some common topically and transdermally delivered substances, highlighting the skin structure and various points of entry
23.2.1 Topical Delivery
The skin is the site of application of numerous cosmetics and cosmeceutical preparations. In this section, we will discuss the importance of targets on active delivery choices. Testing for safety of cosmetic preparations will be discussed in the following sections.
A chemist’s role in overcoming an active’s physicochemical properties to make it suitable for topical and transdermal delivery is a complicated task. Formulators have made use of physicochemical characteristics like molecular weight to stop an active from penetrating through the skin. One such example is a high molecular weight (>500 Da) UVA and UVB absorber Tinosorb® M (bisoctrizole, INCI: methylene bis-benzotriazolyl tetramethylbutylphenol (and) aqua (and) decyl glucoside (and) propylene glycol (and) xanthan gum) developed by Ciba Chemicals (now part of BASF, Ludwigshafen, Germany). Similarly L’Oréal’s sunscreen with Uvinul® N539 (2-ethylhexyl 2-cyano-3,3-diphenyl-2-propenoate, INCI: octocrylene), Parsol® 1789 (avobenzone; INCI: butyl methoxydibenzoylmethane) and Mexoryl® SX (MW-563; ecamsule, INCI: terephthalylidene dicamphor sulfon) was formulated to reduce solar-UV-induced skin damage (Seité et al. 2000). Sunscreens containing ecamsule (Mexoryl® SX) are exclusive to L’Oréal and its brands.
Chlorhexidine is another example of a molecule which binds extensively to the skin surface due to its cationic nature (Judd et al. 2013). This ensures that it does not penetrate into the skin and rather provides for a sustained antimicrobial action when used topically (Karpanen et al. 2008). Insect repellents are another good example of actives that are not intended to penetrate. Penetration retardants and modifiers have been used for this purpose (Kaushik et al. 2010). On the other hand, terbinafine, a highly lipophilic but equally potent antifungal active, with a log P of 5.5, needs a different approach. When formulated as a microemulsion to enhance its stratum corneum solubility using a mixture of surfactant, co-surfactant and oil, terbinafine exhibited enhanced permeation parameters. Microbiological studies of the microemulsion showed better antifungal activity against Candida albicans and Aspergillus flavus compared to marketed products (Baboota et al. 2007). Retinol, with a high molecular weight and lipophilicity, is another active that is used for topical treatment of acne. These unfavourable properties were overcome by delivering Retinol using solid lipid nanoparticles (SLN). The suitability of the SLNs was compared to a nanoemulsion of the same size, with respect to the ability to influence active penetration and distribution within the skin. In both formulations, 500 μg retinol was applied to porcine skin. Following the SLN dispersion for 6 h, a high retinol concentration was found in the stratum corneum and upper epidermis (approximately 3400 ng or 0.68 % in the top layer). The nanoemulsion, however, only transported 2500 ng (0.5 %) into the upper skin strata (Jenning et al. 2000).
For specific disease, conditions where the surface of the skin itself is the target of therapy, formulation and delivery strategy are of greatest importance. Creams, lotions, emulsions and other topical delivery vehicles have been used effectively for this. The challenge here is to modify the formulation according to the active characteristics and skin condition. Being the largest organ, skin is exposed to the damaging effects of UVA and UVB radiation, often leading to erythema. Antioxidants such as Vitamin A, Vitamin E and Vitamin C have shown beneficial effects in reducing oxidative damage to critical cellular components (Trevithick et al. 1993; Stamford 2012). While the photoprotective effect of topical antioxidants applied before UV exposure is well recognized, the beneficial effect of these compounds when administered after irradiation is less obvious (Wu et al. 2012). Only Vitamin A has been proven to be beneficial in treating long-term photoaged and photodamaged skin (Cho et al. 2005). The stratum corneum is a major barrier to the penetration of lipophilic Vitamin E (Cassano et al. 2009), and hydrophilic Vitamin C is prone to excessive oxidation. Oxidation of Vitamin C is triggered by its ionization in aqueous solution (Stamford 2012). Another example where erythema and papular oedema lesions are seen is Herpes labialis. In a recent study, iontophoretic application of 5 % acyclovir cream was tested in a placebo-controlled trial for treatment of herpes, among 200 patients with an incipient cold sore outbreak at the erythema or papular oedema lesion stage. A 20 min iontophoretic treatment cycle shortened the median classic lesion healing time by 35 h in the active group when compared to the control (Morrel et al. 2006). Steroids have also been popularly used in the treatment of erythema and other inflammatory skin conditions (Perry and Trafeli 2009; Paller 2012). Numerous studies have examined various factors controlling percutaneous absorption rates and dermal clearances of steroids (Siddiqui et al. 1989). The lipophilicity of steroids has been associated with their tissue accumulation and toxic effects. However, Magnusson et al. showed that the tissue accumulation of steroids was not linearly related to their lipophilicity, but the binding of steroids to tissue components led to their accumulation (Magnusson et al. 2006). Long-term corticosteroid usage has been associated with skin deterioration, abnormal skin aging and cutaneous mast cell depletion (Lavker and Schechter 1985; Gonzalez and Sethi 2012).
23.2.2 Targeting of Keratinocytes
One of the most widely explored aims of cellular targeting has been in the viable epidermis itself. Inflammatory conditions broadly known as dermatitis or eczema are largely reflected in epidermal involvement (Elias et al. 1999), including spongiosis, acantosis and parakeratosis, as well as some dermal involvement (Freeman-Anderson et al. 2008). The classical treatment for such conditions is the use of topical corticosteroids, which can be classified on a four-point scale of potency (I–IV, mild, moderate, potent, very potent), based on their vasoconstrictor effects in humans. The body site to be treated determines the potency of the steroid to be used. For example, the face should be treated with mild corticosteroids only, such as hydrocortisone and hydrocortisone acetate. Care needs to be taken with the more potent steroids and in the treatment of chronic conditions. Intermittent dosing can be effective in these circumstances.
Salicylic acid has long been used for conditions such as psoriasis and ichthyosis that require keratolytic treatment. At lower concentrations, salicylic acid is known to have keratoplastic effects; however at higher concentrations, it is found to be keratolytic (Gloor and Beier 1984; Schwarb et al. 1999). Coal tar, retinoids and anthralin, as well as corticosteroids are some other commonly used treatments for psoriasis (Mitra and Wu 2010). Psoriatic skin is one such example where the “rigidization” of skin, ascribed to an increase in the levels of cholesterol and fall in the levels of ceramides and the lack of hydration, makes the topical route a big challenge (Wertz et al. 1989). The intricacies of topical delivery into psoriatic skin have lately been addressed by lipoidal carrier systems, such as liposomes. Recent work by Ward et al. describes the delivery of novel immunotherapy using a liposomal platform. Liposomes synthesized to incorporate clodronate were injected intradermally into KC-Tie2 mice. An elimination of the CD11c+, F4 ⁄80+ and CD11b + cells was seen in the skin, and the CD8+ T-cell numbers returned to levels seen in control mice. Further, there was also a marked reduction in the levels of interleukin (IL)-1a, IL-6, IL-23 and tumour necrosis factor (TNFα) (Ward et al. 2011). Peptides and proteins such as elafin and psoriasin are known to be highly upregulated in psoriatic skin (Madsen et al. 1991; Nonomura et al. 1994; Namjoshi et al. 2008). Peptide T analogue, DAPTA, was shown in clinical trials to clear psoriasis lesions and significantly inhibit the monocyte and lymphocyte chemotactic properties of RANTES (a beta chemokine found in increased amounts in psoriatic lesions). A better understanding of the role of these peptides may provide new strategies for the treatment of diseases such as psoriasis (Harder and Schröder 2002).
23.2.3 Targeting of Melanocytes
The synthesis and distribution of melanin contribute to skin and hair colour. However, melanin and pigmentation-associated skin cancers are a major problem, while there is also interest in cosmetic skin whitening treatments where increased epidermal melanin synthesis causes unwanted darkening of the skin. Accurate determination of cutaneous melanin in normal skin and in disease conditions requires the collection of skin biopsies. However, our recent work has used multiphoton tomography and fluorescence lifetime imaging to non-invasively measure levels of epidermal melanin, which can be related to the visible skin colour (Dancik et al. 2013a, b). The use of these techniques has implications for lesion diagnosis and assessment of therapeutic progress.
Related posts:
 Retardation Strategies for Sunscreen Agents
Retardation Strategies for Sunscreen Agents
 Confocal Raman Spectroscopy as a Tool to Investigate the Action of Penetration Enhancers Inside the Skin
Confocal Raman Spectroscopy as a Tool to Investigate the Action of Penetration Enhancers Inside the Skin
 Human Native and Reconstructed Skin Preparations for In Vitro Penetration and Permeation Studies
Human Native and Reconstructed Skin Preparations for In Vitro Penetration and Permeation Studies
 Confocal Microscopy for Visualization of Skin Penetration
Confocal Microscopy for Visualization of Skin Penetration
 Finite and Infinite Dosing
Finite and Infinite Dosing
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


