Chapter 26 Effects of burn Injury on bone and mineral metabolism
 Access the complete reference list online at http://www.expertconsult.com
Access the complete reference list online at http://www.expertconsult.com
 IN THIS CHAPTER
IN THIS CHAPTER  PowerPoint Presentation Online
PowerPoint Presentation Online
Metabolic actions of calcium, phosphate, and magnesium
Calcium
Ca plays a major role in neurotransmission, cell depolarization, impulse propagation, and muscle contractility. In intracellular pathways, after capture by Ca-binding protein or protein kinase C, Ca serves as a second messenger, mediating the secretory release of peptides such as amylase, insulin, and aldosterone. In extracellular metabolism, Ca serves as a cofactor in blood coagulation, specifically in the conversion of prothrombin to thrombin and the activation of several factors in the coagulation cascade.1
Phosphate
PO4 has an integral role in various intracellular processes that involve the storage and transfer of energy. As a component of purine nucleotides such as adenosine-5′-triphosphate (ATP), PO4 groups are exchanged in multiple metabolic reactions that allow energy-demanding activities in the human body. Phosphorylation reactions represent the mainstay of cellular respiration. In the form of phospholipid, PO4 is a major structural component of cell membranes.1
Magnesium
Mg is essential to the cell and especially the mitochondria. As a cofactor, Mg plays a fundamental part in the transfer of PO4 groups. Mg is necessary in reactions requiring ATP or involving replication, transcription, and translation of purine nucleotides.1 Because of its important function in plasma membrane excitability, Mg also serves a role in the stabilization of conditions characterized by abnormal nerve excitation or vasospasm.
Homeostasis of calcium, phosphate, and magnesium
Calcium
Intestinal efficiency to absorb Ca is inversely related to Ca intake. Fractional absorption can be as low as 20% with high Ca intake and as high as 70% with low intake.2 The regulatory mechanism is shown in Figure 26.1. With high Ca intake, transient hypercalcemia occurs, followed by suppression of PTH secretion and PTH-stimulated renal conversion of 25-hydroxyvitamin D to calcitriol (1,25-dihydroxyvitamin D). With low Ca intake the reverse occurs. A transient reduction in serum Ca occurs, followed by a rapid (within 5 minutes) rise in PTH secretion.
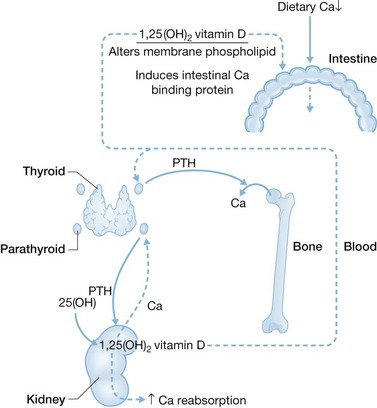
These mechanisms are likely mediated by the parathyroid chief cell Ca-sensing receptor (CaR). The CaR is a membrane-bound G-protein-coupled protein that may be up- or downregulated, in part, by mutations.3 In patients with CaR downregulation, higher circulating Ca is required to suppress PTH production and secretion, giving rise to primary hyperparathyroidism.4 In patients with CaR upregulation less circulating Ca is needed, leading to hypoparathyroidism.5
Intravenous administration of Ca bypasses the intestinal control mechanism and suppresses parathyroid PTH production and kidney calcitriol production. The bone stores 99% of the body’s Ca.1
Phosphate
In contrast to Ca, the intestine plays no significant regulatory role in the absorption of PO4. Approximately 80% of dietary PO4 is absorbed, and the bone stores approximately 90% of the body’s PO4. Homeostatic control appears to rest primarily within the kidney,6,7 and the regulatory mechanisms are likely independent.of PTH.6,7 Thus, the renal excretory rate of PO4 primarily regulates serum PO4 concentrations and maintains them within a normal range. Fibroblast growth factor (FGF)-23 is a key regulator of PO4 and vitamin D metabolism in humans.8 FGF-23 gene mutations cause autosomal dominant hypophosphatemic rickets, a phosphate-wasting disorder. FGF-23-mediated renal phosphate wasting occurs through downregulation of the type II sodium–phosphate co-transporters NPT2a and NPT2c.8
Magnesium
Approximately 60% of the body’s Mg is stored in the skeleton,1 but not at sites where matrix is calcified. Mg absorption varies with dietary intake, with about 40% of an average daily load being absorbed.1 The relationship between Ca and Mg absorption is described as inverse, but the mechanism of this is unclear. Renal excretion is the main route of Mg elimination and it may vary with Mg concentration in serum. A large paracellular pathway for the intestinal absorption and secretion of Mg exists and is dependent on luminal Mg concentrations. The Mg ion channel, TRPM6, is located in intestinal brush border epithelial cells and may participate in Mg homeostasis in the gut. Mg is 70% ultrafiltrable in the serum.1 About 70% of filtered Mg is reabsorbed along the cortical afferent limb of the loop of Henle.1 Hypermagnesemia increases urinary Mg excretion by activating the renal CaR,9 whereas hypomagnesemia increases loop of Henle Mg reabsorption and reduces urinary Mg excretion. Loop diuretics increase urinary Mg excretion. Because little distal tubular Mg reabsorption occurs, an intravenous fluid bolus decreases Mg reabsorption and increases urinary Mg excretion.1
Effect of burn injury on calcium, phosphate, and magnesium homeostasis
Although the effects of burn injury on mineral ions are not fully understood, studies from the University of Texas Medical Branch and Shriners Hospitals for Children in Galveston describe some of the developments in this area. In children with >30% TBSA burns, ionized Ca is, on average, 5% below the lower normal limit.10 In addition, serum PTH levels are too low for ionized Ca levels, indicating hypoparathyroidism. Administration of a standard amount of PTH does not increase urinary cyclic AMP and PO4 excretion,10 pointing to PTH resistance. Mg depletion, encountered in all the burn patients studied,10,11 impairs hypocalcemia-induced PTH secretion and imparts resistance to PTH infusion. The prevalence of Mg depletion may result from resuscitation of patients with Ringer’s lactate, which lacks Mg. Aggressive parenteral Mg supplementation produces repletion in 50% of patients. However, it does not improve hypoparathyroidism,11 making the cause of post-burn hypoparathyroidism unclear. Sheep studies revealed that an approximate 50% upregulation of parathyroid CaR occurs after burn injury,5 which is associated, in humans, with decreased circulating Ca necessary to suppress PTH secretion.3 The proposed mechanism underlying this phenomenon, known as a reduced set point for Ca suppression of PTH secretion, is shown in Figure 26.2. Cytokines, especially IL-1β and IL-6, are highly produced following the systemic inflammatory response. These cytokines stimulate parathyroid cell production of CaR in vitro.12–14
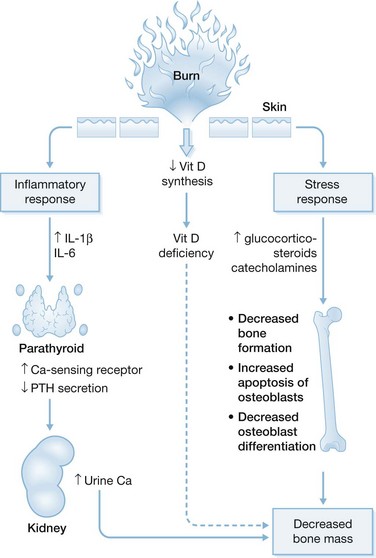
In a study of 11 adult burn patients, serum concentrations PO4, and Mg were low, consistent with abnormalities observed in Ca homeostasis.15 Six patients had low serum ionized Ca concentrations, three of them manifesting hypocalcemia during the first 48 hours after burn. Four were hypophosphatemic; this was most prevalent at post-burn day 7. Five were hypomagnesemic, with this finding most likely to present on post-burn day 3. One patient demonstrated hypercalcemia and one hyperphosphatemia. No patient was hypermagnesemic. Elevated ionized Ca or PO4 was always transient. Studies in adults are not yet as detailed as those in children have been in this regard.
Hypocalcemia cannot be diagnosed from total serum Ca concentration, owing to variability in serum albumin concentrations after burn. Serum ionized Ca concentration yields a more accurate diagnosis. Several mechanisms may underlie hypocalcemia. One is the extracellular–intracellular shift of Ca, supported by Ca accumulation seen in the erythrocytes of a burn patient.16 Another is increased urinary Ca excretion, which occurs in burned children and is consistent with documented secondary hypoparathyroidism.10 Ca loss in tissue exudates could also contribute to hypocalcemia. Although the amount of Ca in wound exudates is likely insufficient to account for post-burn hypocalcemia,17 few studies have measured Ca content in burn wound exudates.
Although fecal Ca losses can be high in burn patients17 and burn-induced increases in endogenous corticosteroids may impair intestinal Ca absorption,18 no evidence suggests that hypocalcemia is caused by corticosteroid-induced impairment of intestinal reabsorption of Ca secreted into the intestinal lumen. Other proposed mechanisms include reduced bone turnover.15,19 However, upregulation of parathyroid CaR by inflammatory cytokines followed by a reduction in set-point for Ca suppression of PTH production remains the most attractive hypothesis.5,10
Studies of 24 children with massive burns demonstrated a low serum concentration of 25-hydroxyvitamin D from as early as 14 months after burn20 up to 7 years,21 correlating with low bone density Z scores.21 Serum 1,25-dihydroxyvitamin D concentrations were normal 2 years after injury, but were low at 7 years in 50% of patients.21 This suggests that these patients became progressively vitamin D deficient.21
Possible explanations for post-burn hypophosphatemia include intracellular PO4 accumulation, inadequate intake, excessive excretion (unlikely in view of documented hypoparathyroidism), or loss into the extravascular fluid. In a review, Dolecek17 found increased urinary PO4 excretion only during the third and fourth weeks after burn in adults, whereas hypophosphatemia occurred earlier. Thus, increased urinary PO4 excretion seen later may be more a function of increased tissue breakdown and filtered load than of inappropriate or excessive urinary PO4 losses. There is little documentation of inadequate PO4 intake following burns. We administer a minimum of 1.6 g PO4 daily in enteral feedings alone.15
The cause of sustained hypomagnesemia after burn is unknown, although excessive urinary and fecal losses occur in adults17 and excessive losses occur in the burn wound.22
Rationale for therapy
Table 26.1 describes treatments for hypocalcemia and hypophosphatemia. Hypocalcemia, especially during the resuscitation effort, can potentiate hypokalemia-induced abnormalities in cardiac muscle23 and block responsiveness to fluid repletion in shock.23 Parenteral Ca during resuscitation does not benefit non-hypocalcemic patients24–26 unless they have hyperkalemia, hypomagnesemia, or Ca channel blocker toxicity.27 Similarly, although caution should be exercised during massive transfusion with citrate-containing blood, Ca therapy may not be necessary in normocalcemic patients and if hepatic and renal functions are minimally impaired. The liver will clear citrate, which may transiently chelate Ca at a rate of 1 unit of blood transfused every 5 minutes.28 Treatment should be initiated only when clinical and electrocardiographic evidence suggests hypocalcemia. Ca infusions should be administered slowly, as rapid Ca replacement can produce cardiac arrhythmias.23,28
| Disturbance | Decision point | Recommended treatment |
|---|---|---|
| Hypocalcemia | Symptomatic | Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree


|