Fig. 27.1
Clinical features of EDSF syndrome. A useful diagnostic clue is the perioral erosions and cheilitis, seen here (a) in a neonate and (b) in a child. Additional features include (c) hypotrichosis or alopecia and (d) palmoplantar keratoderma, typically with painful fissuring
27.3 EDSF Syndrome: Skin and Molecular Pathology
Light microscopy of the skin in EDSF syndrome shows intraepidermal pathology (Fig. 27.2). Typically, there is mild hyperkeratosis and acanthosis with widening of spaces between adjacent keratinocytes, particularly throughout the spinous layer. Ultrastructurally, there are a reduced number of desmosomes that may be small and poorly formed [18]. Although there appears to be acantholysis, the actual plane of weakness is within the desmosomal plaque, i.e. inside the keratinocyte, and thus cleavage in EDSF syndrome could be termed “pseudo-acantholysis”. Skin immunostaining typically shows markedly reduced or completely absent labelling for PKP1, although some residual staining is seen in some cases, depending on the nature of the PKP1 mutation(s) and the antibody probe(s) used. Immunolabelling for other desmosomal proteins is usually of normal intensity, but some redistribution of staining patterns can be seen. Of note, the pattern of desmoplakin labelling tends to be a more cytoplasmic staining with less membranous staining. Likewise, keratin immunolabelling is often compacted in a perinuclear pattern with less staining at the cell periphery close to desmosomes. With regard to the molecular pathology of EDSF syndrome, to date, 14 different pathogenic mutations in the PKP1 gene have been reported, which comprise 3 nonsense, 3 frameshift and 8 splice site mutations (Fig. 27.3). The mutations have been located within the amino terminus of PKP1 as well as the second, third, fourth, seventh, eighth and ninth armadillo-repeat domains of PKP1. In 9 of the 12 published cases that contain molecular studies (lacking in the ref. [13]), the mutations have been homozygous. With respect to genotype-phenotype correlation, nearly all cases involve loss of expression of PKP1, but a slightly milder clinical variant of the syndrome was associated with an in-frame transcript arising from a homozygous donor splice site mutation [9].
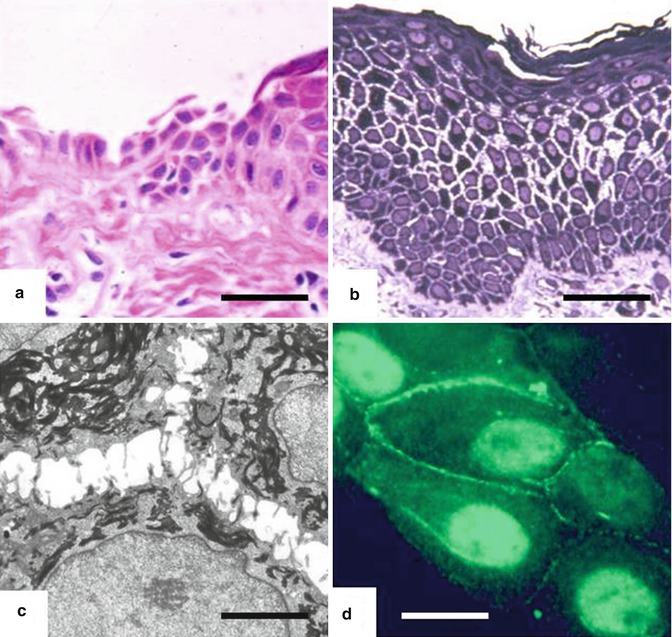
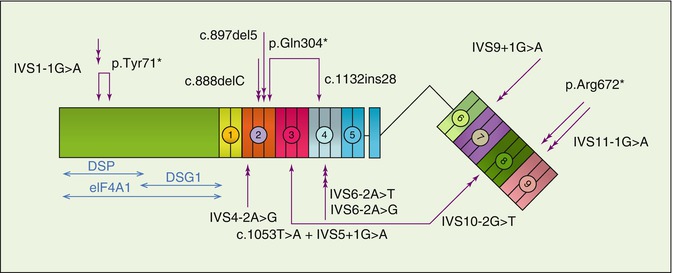
Fig. 27.2
Skin biopsy abnormalities in EDSF syndrome. Light microscopy shows (a) extensive epithelial detachment from the dermis (haematoxylin and eosin; scale bar 50 μ[mu]m), as well as acanthosis and widening of spaces between adjacent keratinocytes throughout the spinous layer (Richardson’s stain; bar = 50 μ[mu]m). Transmission electron microscopy shows (c) keratinocyte separation and retraction of keratin filaments in a compacted perinuclear distribution (bar = 1 μ[mu]m). Immunostaining of normal cultured keratinocytes (d) reveals labelling for PKP1 at the cell peripheries as well as within nuclei (bar = 2 μ[mu]m)
Fig. 27.3
Database of mutations in the PKP1 gene in EDSF syndrome. Double arrows indicate homozygous mutations; joined arrows depict compound heterozygous mutations. Coloured circled numbers (1–9) refer to the armadillo domains of the PKP1 protein. The figure also indicates the protein interaction sites of PKP1 with desmoplakin (DSP), desmoglein 1 (DSG1) and the protein translation initiation factor eIF4A1
27.4 PKP1: Relevance to Human Biology
The discovery of loss-of-function mutations in the PKP1 gene in individuals with EDSF syndrome provides some understanding of the role of PKP1 in human biology, although other studies have provided additional insight. PKP1 is expressed predominantly in suprabasal layers of stratified and complex epithelia, although RT-PCR has revealed that PKP1 is expressed at low levels in most if not all tissues, where it functions independently of desmosomes [19]. Within keratinocytes, PKP1 recruits large amounts of desmoplakin, desmoglein and keratins to the membrane consistent with an in vivo function of increasing desmosome size and strength in suprabasal cells of the skin [20, 21]. However, there are two principal isoforms of PKP1, designated 1a and 1b, generated through alternative splicing of exon 7 [22]. PKP1a is expressed in desmosomes, cytoplasm and nuclei, whereas PKP1b is only expressed in nuclei, where it may interact with single-stranded DNA in vitro (or possibly RNA) and function in protecting cells from DNA damage [23]. The amino terminus of PKP1 binds to desmoplakin and desmoglein 1 [24], but this part of the PKP1 protein also stimulates the recruitment of the translation initiation factor eIF4A1 into the cap complex and also regulates its helicase activity (Fig. 27.3) [25]. Thus PKP1 has a role in protein translation as well as cell adhesion, and its interaction with eIF4A1 offers some insight into mechanisms of epithelial proliferation as well as the association of PKP1 with some malignancies [25]. The link between PKP1 and cancer is not straightforward, however, since loss of PKP1 expression (e.g. by promoter methylation) has been reported in some cancers, such as the transition of Barrett’s oesophagus into adenocarcinoma, in which increased cell motility/migration is thought to contribute [26]. With regard to EDSF syndrome, there does not appear to be any association with cancer susceptibility.
27.5 Summary
The autosomal recessive disorder EDSF syndrome is the first human inherited disorder of desmosomes. It is caused by loss-of-function mutations in the PKP1 gene and approximately 12 cases of this syndrome have been reported. The clinical features of skin erosions, skin crusting and keratoderma with painful fissures implicate PKP1 as an important protein in maintaining desmosomal and epidermal integrity.
References
Related posts:
 Kindlin-1 and Its Role in Kindler Syndrome
Kindlin-1 and Its Role in Kindler Syndrome
 Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
 Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
 Living with Epidermolysis Bullosa: Reviewing the Impact on Individuals’ Quality of Life
Living with Epidermolysis Bullosa: Reviewing the Impact on Individuals’ Quality of Life
 How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


