Dystrophic epidermolysis bullosa (DEB) is relatively well understood. Potential therapies are in development. This article describes the pathogenesis and clinical features of DEB. It also describes therapeutic options and the future of molecular therapies.
Dystrophic epidermolysis bullosa (DEB) is an epidermolysis bullosa (EB) subtype with rather well understood pathogenesis. Its molecular basis—abnormalities of collagen VII—has been known for almost two decades, and, so far, several hundred different mutations in the gene for collagen VII, COL7A1 , have been disclosed in both recessive and dominant forms of DEB (human gene mutation database: www.hgmd.cf.ac.uk/ac/index.php ). Several studies have addressed genotype–phenotype correlations and found the spectrum of biologic and clinical phenotypes to be much broader than initially anticipated. Nevertheless, the cellular and molecular disease mechanisms are reasonably well known and reveal realistic perspectives for the development of biologically valid therapies for DEB in the future.
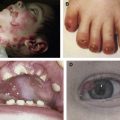
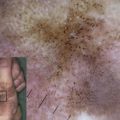
The clinical hallmarks of DEB are trauma-induced blisters and healing with scarring. Dermal–epidermal separation occurs beneath the basement membrane in the uppermost papillary dermis. DEB occurs in all races worldwide and is equally common in boys and girls; its prevalence, however, is not known precisely. Data from the German EB-Network ( www.netzwerk-eb.de ) and the US national EB registry indicate that about 40% of all patients in these databases have DEB. There is some variation in the frequency of the recessive and dominant DEB forms among different populations, however, reflecting each population’s genetic pool.
Molecular pathogenesis of DEB
Blistering in DEB results from structural and functional alterations of the anchoring fibrils (AF), polymers of collagen VII, which anchor the epidermal basement membrane with the dermis. Normal AF are visible in the electron microscope as centrosymmetrically cross-banded fibrils with frayed ends, which emanate from the lamina densa of the basement membrane into the dermal connective tissue. Collagen VII polymerizes in a highly organized manner into the fibrils, which are stabilized by tissue transglutaminase and bind covalently to dermal collagen fibrils, thus securing dermal–epidermal adherence. In DEB skin, collagen VII often is reduced or absent, and electron microscopy reveals paucity, rudimentary structure, or complete lack of the AF.
All DEB forms are caused by mutations in the collagen VII gene, COL7A1 . The spectrum of mutations is broad, and many families have their private mutations. In recessive DEB compound heterozygosity is common (ie, the patient has two different mutations, one inherited from the father and one from the mother). Different combinations of mutations generate a continuum of biologic phenotypes, which explains the varying clinical severity and overlap between different DEB forms.
In most patients with most severe recessive DEB, both COL7A1 mutations cause premature termination codons (PTC), which lead to nonsense mediated mRNA decay, or residual expression of truncated collagen VII polypeptides that are degraded within the cell. As a result, the AF are usually missing in the skin. The genetic background of other recessive DEB forms is heterogeneous, including missense or splice site mutations in COL7A1 in at least one allele. Frequently, the second mutation causes a PTC. These mutation combinations lead to synthesis of defective collagen VII and to structurally abnormal AF. In dominant DEB, heterozygous glycine substitution mutations interfere in a dominant–negative manner with collagen VII synthesis and impair its secretion or the fibrillogenesis of the AF.
Two different mouse models with complete or partial deficiency of collagen VII recapitulate the clinical and morphologic characteristics of recessive DEB in people and have been useful for understanding the molecular pathology of the disease. The collagen VII-knockout mouse exhibits severe blistering that is lethal within about a week after birth. The collagen – hypomorphic mouse, which has about 10% of normal collagen VII levels, lives into adulthood and develops a severe recessive DEB with scarring, nail dystrophy, and mutilating deformities of the paws. Molecular analysis of the extremities of this mouse has revealed that transforming growth factor-beta mediated fibroblast-to-myofibroblast transformation leads to contractile fibrosis, which underlies the mutilating deformities. Both mouse models are also valuable for testing of molecular therapy strategies for DEB.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


