Disorders of the Apocrine Sweat Glands: Introduction
Apocrine glands are adnexal glands that are distributed in the scalp, the axillae, the anogenital region, the eyelids (Moll’s glands), the external auditory meatus (ceruminous glands), and the mammary glands. Apocrine glands can also be found in a more limited distribution on the face and abdomen. Apocrine glands are quiescent until puberty. Embryologically, apocrine glands develop from the upper bulge of the hair follicle late in the fourth month of gestation, with continued development as long as hair follicles develop. A primary epithelial germ (hair germ) grows down from the epidermis and forms an apocrine gland, sebaceous gland, and hair follicle. Apocrine glands are composed of three components: (1) the intraepithelial duct, (2) the intradermal duct, and (3) a coiled gland in the deep dermis or at the junction of the dermis and subcutaneous fat, which contains the secretory portion. The coiled gland consists of one layer of secretory cells around a lumen that is about ten times the diameter of its eccrine counterpart. Contractile myoepithelial cells, a hyaline basement membrane, and connective tissue surround the coiled gland. The predominant mode of apocrine secretion is decapitation, a process where the apical portion of the secretory cell cytoplasm pinches off and enters the lumen of the gland. Apocrine sweat consists mainly of sialomucin. Apocrine sweat is more viscous and produced in much smaller amounts than eccrine sweat (which actually is the wet portion of axillary sweat). The exact function of apocrine glands is unclear, although they are thought to represent scent glands. Their primary sympathetic stimulation is adrenergic.1
Four disorders are considered in this chapter: two primary disorders of the apocrine glands, namely (1) apocrine bromhidrosis and (2) apocrine chromhidrosis; and two secondary disorders, (1) Fox–Fordyce disease and (2) hidradenitis suppurativa (acne inversa), in which apocrine glands become secondarily affected.
Apocrine Bromhidrosis
|
Body odor, osmidrosis, is a common phenomenon in a postpubertal population. Bromhidrosis refers to body odor, which is excessive or particularly unpleasant and apocrine bromhidrosis to such an offensive odor that arises from apocrine glands. It is most often mentioned in the axillae. This condition may contribute to impairment in an individual’s psychosocial functioning. The terminology in the literature is sometimes confusing, using osmidrosis to imply offensive odor, and bromhidrosis to imply osmidrosis in the setting of concomitant hyperhidrosis.2
Disease onset after puberty is common and tends to be more prevalent in African-American populations. There is no geographic predilection, although summer months or warm climates may aggravate the disease. Poor personal hygiene may also be a contributing factor.3
Apocrine secretions are responsible for odor production, primarily through bacterial action on its components. It is accepted that the odorous steroids, the so-called pheromones, among them 16-androstenes, 5α-androstenol, and 5α-androstenone, contribute to osmidrosis.4,5 5α-Reductase type I is expressed in apocrine glands. Individuals with osmidrosis have increased levels of 5α-reductase in their apocrine glands. Because this enzyme catalyzes the conversion of testosterone to 5α-dihydrotestosterone, levels of 5α-dihydrotestosterone may be greater than testosterone in the skin of affected individuals.6 The biotransformation of these steroids is complex and further research is required to delineate these pathways.
Moreover, the axilla hosts many different bacteria, most of which are Gram-positive ones. Bromhidrosis has been particularly associated with the action of aerobic Corynebacterium species. Eccrine bromhidrosis may develop from the action of bacteria on keratin that has been softened by eccrine secretions. In addition, the bacterial action on apocrine secretions produces ammonia and short chain fatty acids. The best-characterized short chain fatty acid is ϵ-3-methyl-2-hexenoic acid.7 This acid is delivered to the surface of the skin on two binding proteins, apocrine-secretion binding proteins (ASOB1 and ASOB2). ASOB2 has been identified as apolipoprotein D.8
The effect of hyperhidrosis (excessive eccrine sweat secretion) on osmidrosis and bromhidrosis is unclear. Some have advocated that excessive eccrine sweat improves apocrine bromhidrosis by flushing away excessive apocrine secretions. Others have postulated that eccrine sweat augments apocrine bromhidrosis by encouraging local spread of apocrine sweat components and enhancing the moist environment in which bacteria flourish.3
Patients complain of an unpleasant body odor. The axillae are the most common affected site, although the genitals or plantar feet may also be affected. The diagnosis is usually clinical. What constitutes a “normal” amount of body odor varies considerably among individuals and ethnic groups. In Asian populations, only slight odor is often considered diagnostic.9,10
Physical examination of the affected individual is usually unremarkable.
There are no associated laboratory abnormalities.
Although some reports do not reveal any abnormalities in the apocrine glands of affected individuals, an increase in the numbers and size of apocrine glands has also been reported.9
Apocrine bromhidrosis should be distinguished from eccrine bromhidrosis, which is far less common (see Chapter 84). Eccrine secretions are distributed in a generalized fashion, are usually odorless, and serve a thermoregulatory function. A plantar location is characteristic for eccrine bromhidrosis. Certain foods (garlic, curry, alcohol), drugs (bromides), toxins, or metabolic causes (disorders of amino acid metabolism) may result in eccrine bromhidrosis (Box 85-1).
|
Frequent washing of the axillae, use of a deodorant or antiperspirant (aluminum chloride), perfumes, and changing of soiled clothing can help. Removal of axillary hair may minimize odor by preventing bacteria and sweat accumulation on the hair shafts. Antibacterial soaps or topical antibacterial agents may also be of benefit.
The injection of botulinum toxin A has been reported to successfully treat genital11 and axillary bromhidrosis.12 The frequency-doubled, Q-switched Nd:YAG laser has also been reported to be an effective noninvasive therapy for axillary bromhidrosis.13
Several surgical measures have been investigated in the treatment of apocrine bromhidrosis. Patient selection is important because surgery is potentially associated with postoperative scar formation, prolonged healing times, infection, and other complications. Upper thoracic sympathectomy has been successful in treating apocrine bromhidrosis either in isolation or in association with palmar hyperhidrosis.2 Surgical removal of the culprit apocrine glands can be achieved either by the removal of subcutaneous tissue in isolation or in combination with axillary skin.14–16 Surgical subcutaneous tissue removal has also been used in association with CO2 laser ablation.17 Although surgical excision may be highly efficacious, depending on the depth of tissue removed and surgical technique used, regeneration and return of apocrine function/osmidrosis and bromhidrosis may develop. Superficial liposuction,18 tumescent superficial liposuction with curettage,19 and ultrasound-assisted liposuction20 have efficacy in the management of apocrine bromhidrosis. In a series of 375 patients, more than 90% reported a satisfactory reduction in odor.20 Another technique with reported efficacy is ultrasonic surgical aspiration of axillary apocrine glands with endoscopic confirmation.21 This technique uses ultrasound to liquefy fat and sweat glands. In contrast, laser hair removal may be associated with intensification of bromhidrosis.22
Apocrine Chromhidrosis
|
Apocrine chromhidrosis is a rare condition characterized by the secretion of colored apocrine sweat. Two variants of apocrine chromhidrosis are recognized: (1) axillary and (2) facial. Involvement of the mammary areola has also been described.23,24 Yonge first recognized facial chromhidrosis in 1709. Shelley and Hurley described this entity in 1954 and associated it with an increased number of lipofuscin granules in apocrine glands.25
Apocrine chromhidrosis is a rare disease. The worldwide prevalence is unknown. Onset of apocrine chromhidrosis is usually at puberty, at the time of increased apocrine gland activity. The disease persists throughout life, improving in the aged. It is reported most commonly in African-Americans.26 Geographic predilections have never been described. Most of the cases reported in the literature involve women; however, there is a lack of sound scientific evidence supporting a female preponderance.
The pigment responsible for causing apocrine chromhidrosis is lipofuscins that are produced in the apocrine secretory cells and excreted to the skin surface. Lipofuscin is a golden-colored pigment that is not specific to apocrine glands. In apocrine chromhidrosis, the lipofuscin granules are in a higher state of oxidation, thereby imparting various colors of pigment, such as yellow, green, blue, or black. Higher states of oxidation produce darker colors.23 It is uncertain why this only develops in some individuals.27
One case of facial chromhidrosis was successfully treated with capsaicin. Nerve endings with receptors for substance P have been found around eccrine sweat glands, suggesting that substance P, a potent vasodilator, may play a role in sweat production and apocrine chromhidrosis.28
Individuals with apocrine chromhidrosis often describe a sensation of warmth, a prickling sensation, or tingling feeling before apocrine gland secretion. Triggers for colored sweating are usually emotional or physical stimuli.26 The morbidity associated with apocrine chromhidrosis stems from the emotional distress experienced by affected individuals.29 Staining of undershirts and handkerchiefs are common complaints.
Individuals with apocrine chromhidrosis develop colored sweat in the axillae or face (Fig. 85-1). Mammary areolar involvement has also been described.23 The pigment produced ranges in color from yellow, blue, green, brown, to even black. The quantity of pigmented sweat produced is usually quite small (approximately 0.001 mL at each follicular orifice).30 The droplets are odorless and dry quickly. Dried secretions appear as dark flecks within affected areas. Axillary involvement causes staining of shirts and undergarments. Facial chromhidrosis commonly develops close to the lower eyelid, including the malar cheeks, and occasionally the forehead.29,30 Colored sweat can also be manually expressed by squeezing in the affected area. Such a maneuver may also be therapeutic.26
Figure 85-1
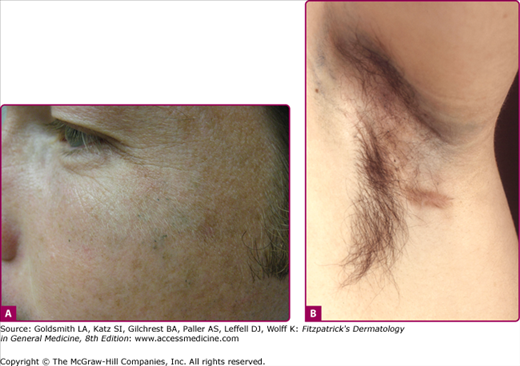
(A) Blue-black sweat produced in a patient with facial apocrine chromhidrosis after gentle squeezing of the cheeks (Reproduced with permission from Chang YC, Anderson N, Soeprono F. Bilateral facial pigmentation. Dermatology Online J. 13(3):16, 2007), (B) blue-black apocrine pigmentation of the axilla and the inflammatory boils of a male patient with hidradenitis suppurativa.
An examination of yellow, blue, or green secretions using a Wood’s light (360 nm) produces a characteristic yellow fluorescence. Black or brown pigment rarely autofluoresces.26 Secretions can be manually expressed if not present at the time of examination. Stained clothing may also fluoresce with Wood’s lamp examination.31 Apocrine glands can be stimulated to produce colored secretions by the injection of epinephrine or oxytocin.
It is reasonable to check a complete blood cell count to exclude a bleeding diathesis, homogentisic levels in urine to exclude alkaptonuria, and bacterial and fungal cultures of affected areas to exclude pseudo-eccrine chromhidrosis.27
The luminal cells of the apocrine sweat glands have an eosinophilic cytoplasm, a large nucleus, and may contain lipofuscin, iron, lipid, or periodic acid-Schiff-positive and diastase-resistant granules.32 Under light microscopy using hematoxylin–eosin staining, an increased number of (yellow–brown) lipofuscin granules may be present in the apical portion of luminal secretory cells of the apocrine glands. The number of granules varies. Additionally, autofluorescence of paraffin-embedded nonstained sections can be demonstrated using a 360-nm wavelength.31 The granules are positive on periodic acid-Schiff stains. Schmorl stain may also be weakly positive.33
Apocrine chromhidrosis must be distinguished from eccrine chromhidrosis (Box 85-2; see Chapter 84). True eccrine chromhidrosis is very rare and occurs when water-soluble pigments are excreted from eccrine glands after the ingestion of certain drugs, such as quinines. Pseudo-eccrine chromhidrosis refers to the development of colored sweat when surface compounds or molecules mix with sweat to produce pigment. A classic example of this type is the formation of blue sweat in copper workers.33 Extrinsic dyes, paints, fungi, and chromogenic bacteria (e.g., Corynebacterium species) are other causes of pseudochromhidrosis.27
|
Adequate therapy for chromhidrosis is lacking. Manual expression of colored secretions may result in a temporary improvement in symptoms for the following 48–72 hours.28 Botulinum toxin type A has reported to be successful in one patient with facial chromhidrosis. This patient experienced a substantial reduction in pigmented sweat and the results were sustained for 4 months.34 Capsaicin is a topical cream that depletes and prevents reaccumulation of substance P levels in unmyelinated, slow-conducting type C sensory fibers. Case reports have demonstrated efficacy of capsaicin in the treatment of facial chromhidrosis.28
Fox–Fordyce Disease
|
Fox–Fordyce disease is an uncommon eruption characterized by pruritic follicular papules that localize to anatomic regions that bear apocrine glands.35 George Henry Fox and John Addison Fordyce originally described it in 1902 in two patients with axillary involvement.36 In 1925, Fischer hypothesized that Fox–Fordyce was a disease of the apocrine glands.37 Shelley and Levy introduced the term apocrine miliaria as a synonym for this disease.38
Approximately 90% of patients with Fox–Fordyce disease are female. Age of onset tends to be after puberty, with most patients between 13 and 35 years of age.38 The incidence of this disorder is unknown. There is no reported ethnic or racial predilection.
The triggers for the development of Fox–Fordyce disease are largely unknown. Shelley and Levy hypothesized that the clinical manifestations of this disease are a result of intraluminal keratin plugging of the follicular infundibula, causing obstruction of the apocrine duct, rupture, and inflammation.38 Although blockage of the apocrine duct seems important in disease development, experimentally plugging the duct has not clinically reproduced disease manifestations.39 One case report detailed the development of Fox–Fordyce disease associated with obstruction of apoeccrine sweat glands.40
Genetics likely plays a role in disease development. This disease has been reported in two patients with Turner syndrome41 and one patient with a small deletion on chromosome 21.42 The disease has also been reported in identical male twins,43 one set of siblings,44 and in father and daughter.45
Hormonal influences on disease have been debated. Disease onset after puberty and improvement with pregnancy and estrogens lends support to a hormonal influence.36,46 However, hormonal studies in one patient with Fox–Fordyce did not reveal any abnormalities.47 Additionally, development before puberty has also been described.48
Patients describe the development of pruritic papules at the time of puberty with gradual worsening. Pruritus can be triggered by emotional excitement or sweating.
Fox–Fordyce disease manifests as numerous symmetrically distributed skin-colored to slightly yellow or red follicular, dome-shaped papules that are equidistant from one another and characteristically intensely pruritic (Fig. 85-2). These papules may resemble lichen planus, lichen nitidus, folliculitis, or syringomas (Box 85-3). Excoriations may be prominent. Apocrine-rich areas are affected, most commonly the axillae. Other areas involved include the pubic area and perineum, mammary areola,7 presternal area, periumbilical area, and upper inner thighs. Only sparse hair growth is seen in the affected area. Apocrine sweat is not produced in affected areas.

Although the clinical features in patients with Fox–Fordyce disease are quite uniform, pathologic findings can vary considerably. The most distinct, relatively consistent pathologic finding is perifollicular foamy macrophages (xanthomatous infiltrates).49 These cells expressed CD68 but lack expression of carcinoembryonic antigen, gross cystic disease fluid protein 15, and periodic acid-Schiff with diastase digestion. Perifollicular mucin, fibrosis, and mast cells in the infiltrate can also be observed. Dilation and hyperkeratosis of the follicular infundibula can also be seen and were previously considered as a histological sign of Fox–Fordyce disease. Other findings include spongiosis and solitary dyskeratotic cells throughout the infundibular epidermis, vacuolar changes at the dermal–epidermal junction in conjunction with some lymphocytes, cornoid lamella in the follicular infundibula with eosinophilic keratinocytes directly below the parakeratotic column, and few lymphocytes in the dermis surrounding the infundibula.35 It has been suggested that transverse histologic sectioning of the specimen is the most effective and efficient method to demonstrate the characteristic pathologic features of Fox–Fordyce disease.50
Treating Fox–Fordyce disease is difficult. Avoidance of excessive sweating or heat may minimize symptoms and flares. Clindamycin with propylene glycol has demonstrated efficacy in small case series in both eliminating symptoms and resolving papules.44,51 Topical tretinoin 0.1%, although potentially irritating, has also demonstrated efficacy.52 Topical pimecrolimus ointment has been shown effective.53 Systemic isotretinoin led to almost complete clearance of lesions, but lesions recurred within 3 months after isotretinoin discontinuation.42 Other reported medical therapies include oral contraceptives,54 testosterone,55 topical or intralesional corticosteroids,56 ultraviolet light,57 and X-rays.47
Electrocoagulation,58 surgical excision,59 and liposuction-assisted curettage60 have all demonstrated efficacy.
Hidradenitis Suppurativa/Acne Inversa
|
Hidradenitis suppurativa/acne inversa (HS) is a rather common, multifactorial, chronic and debilitating inflammatory skin appendage disorder with a notoriously underestimated burden of disease. The first International Hidradenitis Suppurativa Research Symposium (2006, Dessau, Germany) formulated the following definition: HS is a chronic, inflammatory, recurrent, debilitating skin disease (of the hair follicle) that usually presents after puberty with painful, deep-seated, inflamed lesions in the apocrine gland-bearing areas of the body, most commonly the axillary, inguinal, and anogenital regions.61
Conflicting reports have provided a varying assessment of HS epidemiology. A current study recorded an HS prevalence of 1% in a representative sample of the French population (n = 10,000).62 In a previous study, a point prevalence of up to 4.1% based on objective findings, and a 1-year prevalence of 1% based on patient recall was found.63 It seems to be more common in females, with reported female–male ratios ranging from 2:1 to 5:1. The reason for female preponderance is unknown. HS rarely develops before puberty or after menopause, although persistence into menopause is not uncommon.64 The average age of onset is 23 years.65 Although the disease may develop in any apocrine gland-bearing skin, genitofemoral lesions are more prevalent in women, whereas axillary involvement does not demonstrate a gender predilection.63,66,67
HS is a disease of the terminal hair follicle associated with lymphohistiocytic inflammation, granulomatous reactions, sinus tracts, and scarring.61,68 A consistent finding in histological studies of HS is a follicular occlusion due to hyperkeratosis, regardless of disease duration, whereas an isotopic hyperplasia of follicular epithelium is evident.69 This leads to occlusion of the apocrine gland with subsequent follicular rupture, perifollicular inflammation and possible secondary infection, giving rise to clinical findings.69–71 The concept of the follicular occlusion tetrad stems from the concept that HS, acne vulgaris, pilonidal sinus, and dissecting cellulitis share follicular occlusion as an inciting event that eventually leads to disease expression.
Classically, HS was thought to represent a primary disorder of apocrine glands, and was also referred to as apocrinitis.72 The anatomic location of disease in apocrine-bearing skin has supported this concept. Shelley and Cahn provided additional support of this concept by hypothesizing that poral occlusion of the apocrine duct reproduced the clinical and pathologic lesions using their experimental model.73 More recent publications have refuted the concept that this primary event in HS is apocrine gland inflammation and postulate that apocrine glands become secondarily affected.
A family history of HS may be elicited in 26% of patients.74 Several studies have not demonstrated HLA associations.75,76 Others studies have suggested an autosomal dominant mode of inheritance with single gene transmission.77–89 The group of experts, who participated at the first International Symposium has accepted that HS has to be a polygenic disease with sporadic cases having defects in a number of critical genes involved in its pathogenesis and familial cases with probably highly penetrant defect(s) in one of these genes.61 A genome-wide scan in a four-generation Chinese family identified a first locus for HS at chromosome 1p21.1–1q25.3 into a 76-Mb region flanked by the markers D1S248 and D1S2711.90 This locus could not be confirmed by others groups.68,91
In a study of six Han Chinese families with familial disease, frameshift or termination mutations in the γ-secretase complex were identified at 19p13 suggesting that haplosufficiency might play a role in HS in familial cases.92
The coexistence of HS and Crohn disease, particularly with perianal involvement, has been associated with a more fulminant course.80–83 Perianal Crohn disease may be clinically indistinguishable from perianal HS. A pilot study carried on ten HS patients detected no CARD15/NOD2 polymorphisms, found to be associated with Crohn disease.93 This finding adds evidence to the fact that HS has to be distinguished from Crohn disease and its HS-like cutaneous manifestations.94 Other reported associations include pyoderma gangrenosum,84 nephrotic syndrome, and amyloidosis,85 Dowling–Degos disease,86 and arthropathy.87–89 A current systematic literature review reported that follicular occlusion disorders, inflammatory bowel diseases, especially Crohn disease, spondyloarthropathy, other hyperergic diseases, genetic keratin disorders associated with follicular occlusion and squamous cell carcinoma are the most common HS comorbid diseases. A first classification of these major comorbidities and their possible genetic background revealed a list of chromosome loci and genes, which could be HS candidates. Most of these diseases belong to the group of autoinflammatory disorders, where Th17 cell cytokines seem to play a central role.95
The tendency of HS to develop at puberty or postpuberty suggests an androgen influence. Additionally, disease flares have been reported postpartum,86,96 in association with the oral contraception pill, and in the premenstrual period (approximately 50% of patients).97 Antiandrogen therapy has also demonstrated therapeutic benefit in some studies.98–101 However, no biochemical evidence of hyperandrogenism was found in 66 women with HS.102 Additionally, unlike the sebaceous glands, the apocrine glands are not affected by androgens. Thus, the influence of androgens on HS is unclear.
Obesity is unlikely to be a causal factor in HS but is often regarded as an exacerbating factor by increasing shearing forces, occlusion, keratinocyte hydration, and maceration.86,103,104 Obesity may also exacerbate disease by creating a state of androgen excess.77 Weight reduction is recommended for overweight patients and may help to control disease.66 A current multivariate analysis showed a strong association with body mass index [odds ratio = 1.1 (1.1–1.2)].62
The role of bacterial infection in HS is unclear. It is believed that the pathogenic role is indirect, similar to the role of bacteria in acne. Bacterial involvement is thought by some authors to occur secondarily. Routine cultures are often negative, but numerous bacteria have been recovered from lesions. Staphylococcus aureus and coagulase-negative Staphylococci are most frequently isolated.105,106 However, other bacteria, including Streptococci, Gram-negative rods, and anaerobes, have also been identified. These are likely to be colonizing bacteria rather than causative bacteria,86,107 a concept that could explain the increased expression of Toll-like receptor 2 as well as of β-defensin 2 and psoriasin in HS lesions.108,109
The use of tobacco products is more common in HS patients than in healthy controls.110,111 One study determined that 70% of their 43 patient cohort with perineal HS were smokers.112 It is postulated that smoking affects polymorphonuclear cell chemotaxis.64 Multivariate analysis showed a strong association with current smoking (odds ratio = 12.6, 95% CI 8.6–18.4).62 Smoking cessation may improve the clinical course of the disease.
HS is a chronic disease with a variable clinical course.113–115 The diagnosis of HS is clinical, and a biopsy is rarely needed (Box 85-4). Clinical diagnostic criteria are wide and include recurrent disease, scarring, and multifocal location.66
|
One of the most obvious hallmarks of the disease is the restriction to the skin areas affected.113–115 The disease is essentially limited to the intertriginous areas, although aberrant lesions may occur. The sites affected in order of decreasing frequency include: axillary, inguinal, perineal and perianal, mammary and inframammary, buttocks, pubic region, chest, scalp, retroauricular, and eyelid.64 HS is not acne: closed comedones are not seen, since the deep part of the follicle appears to be involved and not its superficial compartments, as seen with acne affecting convex skin surfaces. HS inflammatory lesions are initially transient, but gradually become intransigent and associated with significant scarring.61,115
Three stages of disease are recognized and named after Hurley (I–III).116 In the primary stage, abscesses develop in isolated locations. The secondary stage involves the development of sinus tracts with scars bridging individual lesions. The tertiary stage shows coalescing lesions with scarring and sinus tracts, inflammation, and chronic discharge.
The disease onset is insidious and shows variable severity. Otherwise healthy postpubertal individuals initially may experience slight discomfort or pruritus. After this, a tender papule or deep-seated nodule (0.5 to 2.0 cm) ensues (eFig. 85-2.1). Pustules may also be visualized (eFig. 85-2.2). This nodule may slowly resolve or may expand and coalesce with surrounding nodules to form large painful inflammatory abscesses. These abscesses are rounded without central necrosis and may resolve or rupture spontaneously, producing a purulent discharge (Fig. 85-3). Eventual healing may result in scarring with fibrosis (Fig. 85-4), dermal contractures and rope-like elevation of the skin (Fig. 85-5), and double-ended comedones (eFig. 85-5.1). Sinus tracts may also develop (Fig. 85-6). Sinuses have been reported to involve deep tissue, including muscle and fascia, urethra, and bowel. The process then recurs in an adjacent area or different apocrine-bearing site.66
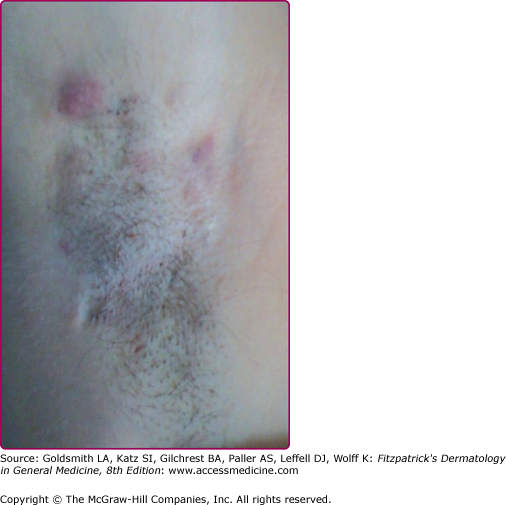
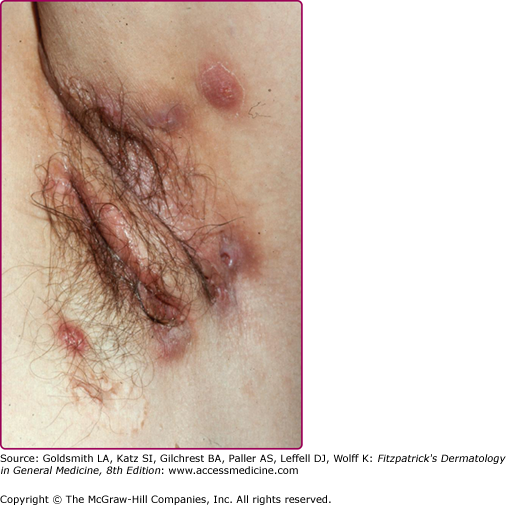
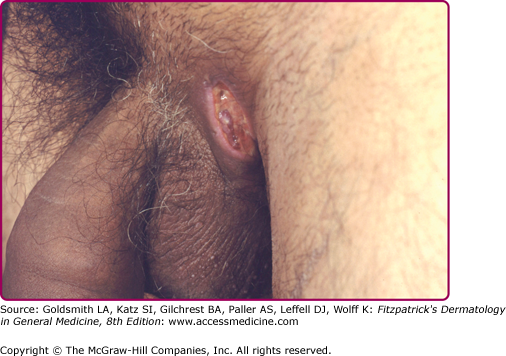
Figure 85-4
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


