Introduction282
INTRODUCTION
The pigmentary system
DISORDERS CHARACTERIZED BY HYPOPIGMENTATION
PIEBALDISM
Note: Some of the associated abnormalities (e.g. Hirschsprung’s disease) were described prior to accurate genetic knowledge. They may have been assigned to the wrong subgroup.
PBT = piebald trait; WS = Waardenburg’s syndrome.
Disease
OMIM
Gene symbol
Gene locus
Comments
PBT
172800
KIT
SNA12
4q11–q12
8q11
Two loci. White forelock, absence of pigment on forehead, chin, chest, abdomen, and extremities
WS type I
193500
PAX3
2q35
WS type IIa
193510
MITF
3p14.1–p12.3
WS type IIb
600193
1p21–p13.3
WS type IIc
606662
8p23
WS type IId
608890
SLUG (SNA12)
8q11
WS type IIe
611584
SOX10
22q13
WS type III
(Klein–Waardenburg)
148820
PAX3
2q35
Is allelic to WS I or it involves a contiguous gene. Upper limb abnormalities
WS type IV
(Waardenburg–Shah)
277580
EDNRB
EDN3
SOX10
20q13.2–q13.3
13q22
22q13
Hirschsprung’s disease. May involve endothelin-B receptor (EDNRB) or its ligand endothelin-3 (EDN3)
Histopathology
VITILIGO
Pathogenesis
Treatment of vitiligo
Histopathology
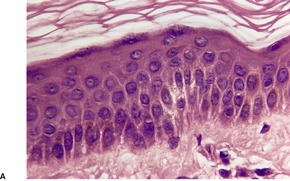
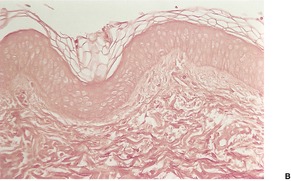
Fig. 10.1
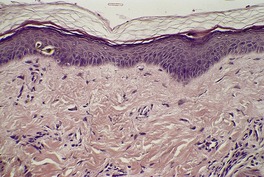
Fig. 10.2
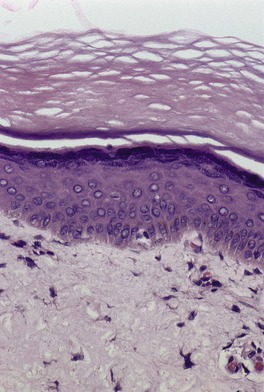
Fig. 10.3
Electron microscopy
OCULOCUTANEOUS ALBINISM
Hermansky–Pudlak syndrome and Chédiak–Higashi syndrome are other variants.
Type
OMIM
Gene symbol
Gene name
Gene locus
Comments
OCA1A
203100
TYR
Tyrosinase
11q14–q21
Complete absence of tyrosinase activity in melanocytes
OCA1B
606952
TYR
Tyrosinase
11q14–q21
Reduced activity of tyrosinase; splicing mutation described
OCA2
203200
P (OCA2)
Pink-eyed dilution
15q11–q13
P gene also deleted in cases of Prader–Willi and Angelman syndrome; variant in Africa with palmoplantar freckles
OCA3
203290
TYRP1
Tyrosinase-related protein 1
9p23
Found predominantly in Africans
OCA4
606574
MATP (SLC45A2)
Membrane-associated transporter protein
5p13.3
Common in Japan
Type 1A oculocutaneous albinism (OCA1A)
Yellow mutant oculocutaneous albinism (type 1B; OCA1B)
Oculocutaneous albinism type 2 (OCA2)
Tyrosinase-positive oculocutaneous albinism (OCA2 variant)
Oculocutaneous albinism type 3 (OCA3)
Oculocutaneous albinism type 4 (OCA4)
Hermansky–Pudlak syndrome
Histopathology
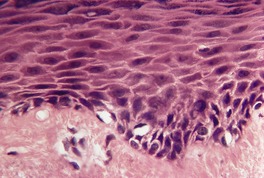
Fig. 10.4
Electron microscopy
CHÉDIAK–HIGASHI SYNDROME
Histopathology
Electron microscopy
GRISCELLI SYNDROME
Histopathology
Electron microscopy
ELEJALDE SYNDROME
Histopathology
PROGRESSIVE MACULAR HYPOMELANOSIS
Histopathology323
Electron microscopy
TUBEROUS SCLEROSIS (‘ASH LEAF SPOTS’)
Histopathology
Electron microscopy6. and 334.
IDIOPATHIC GUTTATE HYPOMELANOSIS
Histopathology337.338. and 342.
HYPOMELANOSIS OF ITO
Histopathology
Electron microscopy
NEVUS DEPIGMENTOSUS
Histopathology
Electron microscopy334
PITYRIASIS ALBA
Histopathology
POSTINFLAMMATORY LEUKODERMA
Histopathology
NEVUS ANEMICUS
Histopathology
DISORDERS CHARACTERIZED BY HYPERPIGMENTATION
GENERALIZED HYPERPIGMENTARY DISORDERS
Histopathology
UNIVERSAL ACQUIRED MELANOSIS
Histopathology
ACROMELANOSIS
Histopathology
FAMILIAL PROGRESSIVE HYPERPIGMENTATION
Histopathology433
IDIOPATHIC ERUPTIVE MACULAR PIGMENTATION
Histopathology
DYSCHROMATOSIS SYMMETRICA HEREDITARIA
Histopathology
DYSCHROMATOSIS UNIVERSALIS HEREDITARIA
Histopathology
PATTERNED HYPERMELANOSIS
Histopathology
FAMILIAL GIGANTIC MELANOCYTOSIS
Histopathology492
CHIMERISM
Histopathology
MELASMA
Histopathology
ACQUIRED BRACHIAL DYSCHROMATOSIS
Histopathology514
EPHELIS (FRECKLE)
Histopathology
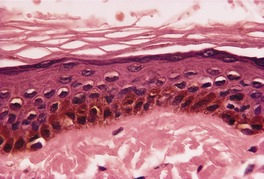
Fig. 10.5
CAFÉ-AU-LAIT SPOTS
Histopathology
MACULES OF ALBRIGHT’S SYNDROME
Histopathology
LAUGIER–HUNZIKER SYNDROME
Histopathology
PEUTZ–JEGHERS SYNDROME
BANNAYAN–RILEY–RUVALCABA SYNDROME
BECKER’S NEVUS
Histopathology
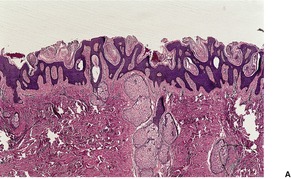
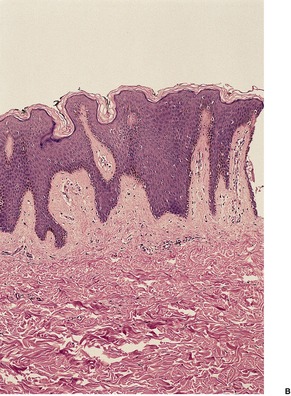
Fig. 10.6
Electron microscopy
DOWLING–DEGOS DISEASE
Histopathology600. and 610.
Electron microscopy
POSTINFLAMMATORY MELANOSIS
Histopathology
PRURIGO PIGMENTOSA
Histopathology656.658.659.660.675. and 676.
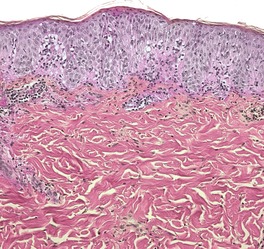
Fig. 10.7
GENERALIZED MELANOSIS IN MALIGNANT MELANOMA
Histopathology678
DERMATOPATHIA PIGMENTOSA RETICULARIS
NAEGELI–FRANCESCHETTI–JADASSOHN SYNDROME
INCONTINENTIA PIGMENTI
Histopathology
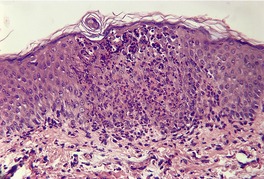
Fig. 10.8
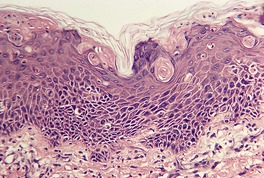
Fig. 10.9
RETICULATE PIGMENTARY DISORDER WITH SYSTEMIC MANIFESTATIONS
Histopathology
FRICTIONAL MELANOSIS
NOTALGIA PARESTHETICA
Histopathology
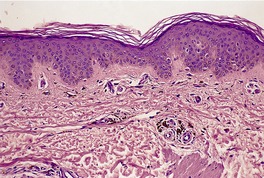
Fig. 10.10
‘RIPPLE’ PIGMENTATION OF THE NECK
‘TERRA FIRMA-FORME’ DERMATOSIS
Histopathology
Disorders of pigmentation
Disorders characterized by hypopigmentation282
Piebaldism282
Vitiligo283
Oculocutaneous albinism286
Type 1A oculocutaneous albinism (OCA1A)286
Yellow mutant oculocutaneous albinism (type 1B; OCA1B)286
Oculocutaneous albinism type 2 (OCA2)286
Tyrosinase-positive oculocutaneous albinism (OCA2 variant)286
Oculocutaneous albinism type 3 (OCA3)287
Oculocutaneous albinism type 4 (OCA4)287
Hermansky–Pudlak syndrome287
Chédiak–Higashi syndrome287
Griscelli syndrome288
Elejalde syndrome288
Progressive macular hypomelanosis288
Tuberous sclerosis (‘ash leaf spots’)288
Idiopathic guttate hypomelanosis289
Hypomelanosis of Ito289
Nevus depigmentosus289
Pityriasis alba289
Postinflammatory leukoderma290
Nevus anemicus290
Disorders characterized by hyperpigmentation290
Generalized hyperpigmentary disorders291
Universal acquired melanosis291
Acromelanosis291
Familial progressive hyperpigmentation291
Idiopathic eruptive macular pigmentation291
Dyschromatosis symmetrica hereditaria292
Dyschromatosis universalis hereditaria292
Patterned hypermelanosis292
Familial gigantic melanocytosis292
Chimerism293
Melasma293
Acquired brachial dyschromatosis293
Ephelis (freckle)293
Café-au-lait spots293
Macules of Albright’s syndrome294
Laugier–Hunziker syndrome294
Peutz–Jeghers syndrome294
Bannayan–Riley–Ruvalcaba syndrome294
Becker’s nevus294
Dowling–Degos disease295
Postinflammatory melanosis296
Prurigo pigmentosa296
Generalized melanosis in malignant melanoma296
Dermatopathia pigmentosa reticularis297
Naegeli–Franceschetti–Jadassohn syndrome297
Incontinentia pigmenti297
Reticulate pigmentary disorder with systemic manifestations298
Frictional melanosis298
Notalgia paresthetica298
‘Ripple’ pigmentation of the neck299
‘Terra firma-forme’ dermatosis299
This chapter deals with the various disorders of cutaneous pigmentation, excluding those entities in which there is an obvious lentiginous proliferation of melanocytes in sections stained with hematoxylin and eosin; it also excludes tumors of the nevus-cell–melanocyte system. Both of the excluded categories are discussed in Chapter 32. Cutaneous pigmentation may also result from the deposition of drug complexes in the dermis. This category of pigmentation is discussed among other cutaneous deposits in Chapter 14. A related condition is the excessive dietary intake of carotenoid-containing foods, which may cause yellow-orange discoloration of the skin.1. and 2.
Cutaneous pigmentary disorders can be divided into two major categories: disorders with hypopigmentation and those with hyperpigmentation. The dyschromatoses, in which areas both of hypopigmentation and hyperpigmentation are present, have been arbitrarily included with the disorders of hyperpigmentation. A detailed list of all conditions resulting in discolorations of the skin was published in 2007. 3
The pigmentary system involves a complex set of reactions with numerous potential sites for dysfunction.4. and 5. Melanin is produced in melanosomes in the cytoplasm of melanocytes by the action of tyrosinase on tyrosine. A number of intermediate steps involving the formation of dopa and dopaquinone take place prior to the synthesis of melanin. The melanin synthesized in any one melanocyte is then transferred to an average of 36 keratinocytes by the phagocytosis of the melanin-laden dendritic tips of the melanocytes. 6 The protease-activated receptor 2 (PAR-2), which is expressed on keratinocytes, is a key receptor involved in melanosome transfer. 7 Other important participants in this process include kinesin and actin-associated myosin V. The latter protein secures the peripheral melanosomes, preparing them for transfer to surrounding keratinocytes. 8 This transfer of melanin can be disrupted by any inflammatory process involving the basal layer of the epidermis. Specific enzyme defects and destruction of melanocytes are other theoretical causes of hypopigmentation.
The pathogenesis of hyperpigmentation is not as well understood. Prominent pigment incontinence is an obvious cause of hyperpigmentation. Ultrastructural examination in some disorders of hyperpigmentation has shown an increase in size or melanization of the melanosomes, although in others the reasons for the basal hyperpigmentation have not been determined.
Skin color in the various races is an interesting topic, but beyond the scope of this book. A historical perspective was published recently. 9 Another paper has reviewed various aspects of racial and ethnic groups with pigmented skin, now called skin of color. 10
The disorders of hypopigmentation will be discussed first.
There are multiple potential sites for dysfunction in the formation of melanin pigment in basal melanocytes. 6 Attempts have been made to categorize the various diseases with hypopigmentation on the basis of their presumed pathogenesis. The following categories may be considered.
1. Abnormal migration/differentiation of melanoblasts: piebaldism, Waardenburg’s and Woolf’s syndromes.
2. Destruction of melanocytes: vitiligo, Vogt–Koyanagi–Harada syndrome, chemical leukoderma.
3. Reduced tyrosinase activity: oculocutaneous albinism type 1A, phenylketonuria(?).
4. Abnormal structure of melanosomes: ‘ash leaf spots’ of tuberous sclerosis, Chédiak–Higashi syndrome, progressive macular hypomelanosis.
5. Reduced melanization and/or numbers of melanosomes: albinism (other tyrosinase-positive variants), Griscelli syndrome, Elejalde syndrome, idiopathic guttate hypomelanosis, hypomelanosis of Ito, ‘ash leaf spots’, pityriasis versicolor (tinea versicolor), nevus depigmentosus.
6. Reduced transfer to keratinocytes: nevus depigmentosus, pityriasis alba, postinflammatory leukoderma, pityriasis versicolor (tinea versicolor), Chédiak–Higashi syndrome. Increased degradation of melanosomes within melanocytes may also apply in some conditions listed in this section.
7. Abnormal vasculature: nevus anemicus.
In addition to the conditions listed above, there are isolated reports of one or more cases in which the hypopigmentation does not correspond neatly to any of the named diseases.11.12. and 13. These cases will not be considered further.
Phenylketonuria, an autosomal recessive disorder with a deficiency of the enzyme L-phenylalanine hydroxylase, is characterized by oculocutaneous pigmentary dilution in addition to neurological abnormalities.6. and 14. There are several steps in the biosynthesis of melanin that may be affected by this enzyme deficiency. As biopsies are rarely taken, this condition will not be discussed further.
In piebaldism (partial albinism – OMIM 172800), an autosomal dominant disorder, there are non-progressive, discrete patches of leukoderma present from birth.6. and 15. The chalk-white areas of hypomelanosis involve the anterior part of the trunk, the mid-region of the extremities, the forehead, and the mid-frontal area of the scalp beneath a white forelock. 6 This hair change is present in up to 90% of those with piebaldism and it is sometimes found as an isolated change in the absence of cutaneous leukoderma. 16 Regression of the white forelock has also been reported. 17 Within the areas of hypomelanosis there are hyperpigmented and normally pigmented macules of various sizes. 18
There are several rare syndromes in which extracutaneous manifestations accompany the piebaldism.6.15.19.20. and 21. Examples include the various types of Waardenburg’s syndrome, in which piebaldism is associated with neurosensory hearing loss and other abnormalities including Hirschsprung’s disease.22.23.24.25.26.27. and 28. Four major variants of Waardenburg’s syndrome have now been described, each one due to the involvement of different genes. The various types are listed in Table 10.1. PAX3, the gene responsible for type I, regulates MITF, the gene responsible for type II. The MITF gene (microphthalmia-associated transcription factor) is assigned to chromosome 3p14.1–p12.3. 29 When type IV Waardenburg syndrome (OMIM 277580) is due to a mutation in the SOX10 gene it usually, but not always, is associated with Hirschsprung’s disease.30. and 31. Neurofibromatosis 1 (NF-1) has also been associated with piebaldism.32. and 33. Poliosis has also followed herpes zoster in the same dermatome; this may be an example of Wolf’s isotopic response. 34
Above features + dystopia canthorum
Cochlear deafness ± Hirschsprung’s disease
Pigmentary changes, absent dystopia canthorum (major difference from type I)
May have hearing loss
Hirschsprung’s disease reported
Piebaldism results from mutations of the KIT proto-oncogene, which encodes a cell-surface receptor, tyrosine kinase, whose ligand is the stem/mast cell growth factor.35. and 36. In humans, the KIT proto-oncogene has been mapped to the proximal long arm of chromosome 4 (4q11–q12).21.36. and 37. Some cases of piebaldism are caused by a mutation in the gene encoding the zinc finger transcription factor (SNA12) located on chromosome 8q11. 38 Variations in the phenotype relate to the site of the KIT gene mutation. A novel KIT mutation, Val620Ala, results in piebaldism with progressive depigmentation. 39 In mice, KIT-mediated signal transduction is required in embryogenesis for the proliferation and migration of melanoblasts from the neural crest. It appears to be required, in humans, for melanocyte proliferation.16.35. and 36. The successful use of autologous grafts to repigment the affected areas is not inconsistent with these theories.40. and 41.
There are usually no melanocytes and no melanin in the leukodermic areas. Sometimes a small number of morphologically abnormal melanocytes are present, particularly near the margins of hypopigmentation. These melanocytes may have spherical melanosomes. Some clear cells, representing Langerhans cells, are usually present in the epidermis. 42
The hyperpigmented islands contain normal numbers of melanocytes: there are abundant melanosomes in the melanocytes and in keratinocytes. There are no dopa-positive melanocytes in the hair bulbs of the white forelock. 23
Vitiligo (OMIM 193200) is an acquired, idiopathic disorder in which there are depigmented macules of variable size which enlarge and coalesce to form extensive areas of leukoderma.43.44.45. and 46. It results from selective destruction of melanocytes. 47 An erythematous border is occasionally present in the initial stages.6.48.49.50. and 51. Repigmentation may lead to several shades of color in a particular lesion, 52 as may transitional stages in depigmentation (trichrome vitiligo). 53 The incidence in white people is approximately 1%, 54 but a study from China has shown that this figure is an overestimation of its incidence. 55 Its first description dates back more than 3000 years. 56 This condition may develop at any age, although in 50% or more of affected persons it appears before the age of 20 years.57.58.59.60. and 61. If there is an extended family history of vitiligo, onset is likely to be at an earlier age. 62 Late-onset vitiligo has been a neglected entity. 63 Almost 15% of late-onset cases demonstrate the Koebner phenomenon; 63 it may occur at any age. 64 A family history is present in up to 25% of cases; the inheritance appears to be polygenic.44.65.66.67. and 68. High-risk haplotypes have been identified in some groups;47. and 69. HLA-A2 is one of these. 70 The angiotensin-converting enzyme (ACE) gene has an association with the development of vitiligo. 71 Vitiligo does not appear to be caused by mutations in the GTP-cyclohydrolase I gene, which regulates melanin biosynthesis. 72
There is a predilection for the face, back of the hands, axillae, groins, umbilicus, and genitalia, and for the skin overlying bony areas such as the knees and elbows. 44 Vitiligo has been reported on the anterior neck in Muslim women. 73 It is thought to be due to the Koebner phenomenon resulting from the wearing of scarves which are tied with metallic or plastic pins in this region. 73 Periocular vitiligo commenced in one patient around a congenital divided nevus of the eyelid. 74 Sometimes the depigmented area is segmental or dermatomal in distribution (type B); 60 more often it is more generalized (type A).75.76. and 77. Universal vitiligo in which the entire body is affected is rare. 78 Repigmentation seldom occurs in type B, which is also resistant to treatment and more common in children.76.79.80. and 81.
Approximately 20–30% of individuals with vitiligo (usually those with bilateral/generalized disease) 79 have an associated autoimmune and/or endocrine disorder6.71.82. and 83. such as Hashimoto’s disease,84. and 85. hyperthyroidism, pernicious anemia, Addison’s disease, 86 insulin-dependent diabetes mellitus,87.88. and 89. and alopecia areata.59.90. and 91. Less frequent associations include various lymphoproliferative diseases, 92 morphea, 93 chronic actinic dermatitis, 94 urticaria, 60 pemphigus vulgaris, 95 the mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes syndrome (MELAS), 96 Crohn’s disease,97. and 98. autoimmune polyglandular syndrome,99. and 100. prior infection with cytomegalovirus,101.102. and 103. HIV infection, 104 idiopathic CD4+ T-cell lymphocytopenia, 105 and chronic mucocutaneous candidosis. 106 The reported association of vitiligo with psoriasis and erythema dyschromicum perstans107 is probably fortuitous. Depigmentation resembling vitiligo has been reported following contact with hydroquinones, certain phenolic agents, 44 cinnamic aldehyde in toothpaste, 108 topical minoxidil, 109 flutamide, 110 chloroquine, 111 imiquimod,112.113. and 114. tazarotene, 115 adalimumab, 116 and PUVA therapy.117.118. and 119. Ganciclovir, used in the treatment of graft-versus-host disease (GVHD), produced extensive vitiligo in one patient. 120 Vitiligo has also followed the use of interferon-α therapy for hepatitis B infection. 121 It has also occurred at the site of interferon-α-2b injection in a patient with chronic viral hepatitis C. 122 Capecitabine has produced hyperpigmentation in non-vitiliginous skin in a patient with cancer treated with this drug. 123 Many cases of depigmentation, some resembling vitiligo, have been reported following the use of the tyrosine kinase inhibitor imatinib, used in the treatment of chronic myeloid leukemia.124.125.126. and 127.
Vitiligo may be accompanied by a variety of ocular pigmentary disturbances. 128 The best known of these is the Vogt–Koyanagi–Harada syndrome, which includes uveitis, poliosis, dysacusis, alopecia, 129 vitiligo, and signs of meningeal irritation.130. and 131. Not all these features are present in all cases. An immunological etiology has been suggested. 132 A rare variant of this syndrome with inflammatory vitiligo has been described. 133
Sometimes there is a history in the patient or the patient’s immediate family of premature graying of the hair (poliosis), a halo nevus, or even a malignant melanoma.77. and 134. Vitiligo may be a presenting sign of metastatic melanoma. 135 It is interesting to note that individuals with metastatic melanoma who develop vitiligo-like depigmentation have a better prognosis than those who do not.136. and 137. Both lesions have clonally expanded T cells with identical BV (β variable) regions. 138
The onset of vitiligo is usually insidious with no precipitating cause. In approximately 20% of cases it develops after severe sunburn or some severe emotional or physical stress.44. and 84. The majority of cases have a progressive clinical course.139. and 140. In generalized forms (vitiligo vulgaris) the depigmentation may eventually involve large areas of skin. Some repigmentation may occur but it is usually incomplete and short lived.44. and 130. Repigmentation probably involves melanocytes from hair follicles (perifollicular repigmentation).141. and 142. It may also occur from melanocytes in adjacent normal skin (marginal repigmentation) and possibly from dopa-negative melanocytes in vitiliginous skin that have been hypothesized to give diffuse repigmentation. 143 Marginal and perifollicular repigmentation are more stable than the diffuse form, which tends to result when steroid therapy is used. 143 Eventually the process of depigmentation ceases. The evolution and therapeutic monitoring of vitiligo can be carried out using in-vivo reflectance confocal microscopy. 54 Vitiligo may have a significant effect on the psychological well-being of some patients, 144 particularly dark-skinned persons, in whom the lesions may be confused with leprosy, resulting in social stigmatization.61. and 145.
Several hypotheses have been proposed to explain the destruction of melanocytes which results in the depigmentation.46.130.146. and 147. These may be summarized as the neural, the self-destructive, the inherent defect, and the autoimmune theories. 148 They are not mutually exclusive.86. and 149. A recent study provides evidence for a link between the neural and apoptotic pathways in the pathogenesis of the disease. 150 The neural hypothesis suggests that a neurochemical mediator released at nerve endings results in destruction of melanocytes. It has been proposed that the segmental form (type B) of vitiligo results from dysfunction of sympathetic nerves in the affected areas. 76 Support for this hypothesis comes from the finding of increased neuropeptide Y activity in vitiligo. 151 The self-destruction hypothesis (autocytotoxicity) is based on the known toxicity of melanin precursors for melanocytes. It is assumed that affected individuals have an intrinsic inability to eliminate or handle these toxic precursors, such as free radicals, which accumulate and result in the destruction of melanocytes by apoptosis.45. and 152. More recent studies have confirmed that oxidative stress is involved in the pathophysiology of vitiligo.153. and 154. Whether such mechanisms are involved in producing the DNA damage observed in patients with vitiligo is unknown. 155 Experimental studies suggest that early cell death of vitiligo melanocytes is related to their increased sensitivity to oxidative stress, which may in some way be linked to the abnormal expression of tyrosinase-related protein (TRP-1). 156 The autoimmune hypothesis, which is currently most favored, particularly for the generalized forms, proposes that antibody-dependent, cell-mediated cytotoxicity utilizing natural killer cells is responsible for the loss of melanocytes.157. and 158. Type 1 cytokines have been incriminated in this process. 159 Infiltrating T cells and macrophages have been observed adjacent to the remaining perilesional melanocytes in generalized vitiligo. 160 However, other studies have suggested that antibodies to melanocytes in the IgG fraction of patients’ serum may be the effector mechanism for melanocyte destruction;82.161.162.163. and 164. the level of the antibodies correlates with disease activity. 165 The antibodies appear to be directed against tyrosinase in some patients, despite earlier reports to the contrary.166.167.168. and 169. At least 15 different antigens may be recognized in some individuals with vitiligo by vitiligo autoantibodies. 82 Other experiments also downplay the role of natural killer and lymphokine-activated killer cells in the pathogenesis.170.171. and 172. Various other abnormalities in the immune system have been recorded in vitiligo. 173 These include a decrease in T-helper cells,174.175. and 176. an increase in natural killer cells,175. and 177. circulating antibodies to surface antigens on melanocytes178.179.180. and 181. and to certain melanoma cell lines,182. and 183. aberrant expression of complement regulatory proteins, 184 an increase in met-enkephalin secretion, 185 a decrease in the expression of c-kit protein by melanocytes adjacent to lesional skin,186. and 187. abnormal expression of MHC class II molecules and ICAM-1 by perilesional melanocytes, 188 and possible functional impairment of Langerhans cells.189. and 190. In melanoma-associated vitiligo, CD8+ T cells are important in the pathogenesis. 191
Vitiligo has developed in a patient after bone marrow transplantation from a donor with vitiligo. 192 It has also developed in two patients who were given a donor lymphocyte infusion for leukemia relapse over 3 years after bone marrow transplantation. 193
Recent work has associated the cytotoxic T lymphocyte antigen-4 (CTLA-4) gene product, which is involved in controlling T-cell apoptosis, with susceptibility to autoimmune diseases including vitiligo. It is important to note that significant associations of vitiligo with CLTA-4 polymorphic markers are only seen in patients with concomitant autoimmune diseases, suggesting there may be two forms of vitiligo. 82 A more recent paper concluded that CTLA-4 was not associated with a risk of generalized vitiligo, but that polymorphisms in the PTPN22 gene were. 194 Polymorphisms in the autoimmune regulator gene (AIRE) are sometimes found in patients with vitiligo. 195
Treatment options for vitiligo depend predominantly on the body surface area (BSA) involved, and to a lesser extent the site (particularly if it is the face), and the age of the patient. If less than 10% of the BSA is involved various treatments can be used including topical corticosteroids, topical calcineurin inhibitors, phototherapy, and transplantation with autologous melanocytes or epidermal cell suspensions (see below).196.197.198.199.200.201.202.203.204. and 205. When larger areas of the body are involved (BSA 11–80%) phototherapy with or without antioxidants is the treatment of choice.205.206.207. and 208. Some success has been achieved in generalized vitiligo with autologous minigrafting.199.209.210.211.212.213. and 214. If more than 80% of the BSA is involved, consideration should be given to depigmenting the normal skin with a hydroquinone cream or a combination of the cream with Q-switched ruby laser therapy.205. and 215.
With respect to topical therapy, moderate to high potency topical corticosteroids are efficacious in children, but their use may be associated with some systemic absorption. 216 Topical tacrolimus ointment is an effective alternative therapy for childhood vitiligo, particularly involving the head and neck. 217 Rapid enlargement of a malignant melanoma occurred in a child with vitiligo after the application of topical tacrolimus. 218 Pimecrolimus cream has been used for eyebrow/eyelid and genital lesions. 219 Topical calcipotriol has not proved effective, 220 but repigmentation was achieved in a young girl using 0.0002% tacalcitol cream and sunlight exposure. 221 Tanning preparations, topical dyes, and tattooing have been used for small patches but they do not always produce an ideal color match. 205 Effective camouflaging of vitiligo can be achieved with 6% dihydroxyacetone cream. It is a component of self-tanning agents. 222 It may be less expensive than the widely used cosmetic camouflages, such as Dermablend® and Covermark®. 222 As the accumulation of hydrogen peroxide (H2O2) has been documented in vitiligo, a recent trial using low-dose, narrowband, UVB-activated pseudocatalase PC-KUS to remove/reduce H2O2 levels in vitiligo was carried out. 223 Improvement occurred in a significant number of patients. 223 Proton pump inhibitors have produced repigmentation, but vitiligo has also been precipitated by these drugs. 224 Repigmentation of vitiligo has occurred during efalizumab therapy for psoriasis. The vitiligo reappeared on cessation of the drug. 225
In the case of phototherapy, recent trials have shown promising results with narrowband ultraviolet-B radiation (NB-UVB), which appears to give better results than PUVA and related therapies.206.207.208.226. and 227. Certain body sites, such as the face, respond better with this treatment. 228 Even better results have been achieved recently with the 308 nm monochromatic excimer light, although more trials are needed.229.230. and 231. It induces repigmentation more rapidly. If NB-UVB phototherapy is combined with supplemental antioxidants, such as α-lipoic acid or oral Polypodium leucotomos, the clinical effectiveness is enhanced.232. and 233. With phototherapy a perifollicular pattern of repigmentation is the predominant one; it is the most stable pattern of repigmentation. 143 PUVA may also have a stimulating effect on melanocytes, not only locally, but also systemically. This may be mediated by endothelin-1, which is significantly elevated after this therapy. 234 PUVA keratoses have developed in vitiligo skin treated with PUVA therapy. 235
Several surgical procedures have been used to treat vitiligo which is refractory to medical treatment.236. and 237. Techniques using non-cultured autologous melanocyte or epidermal cell suspensions offer an advantage over earlier grafting techniques, allowing treatment of a larger recipient area with a smaller donor site.238.239. and 240. Repigmentation of vitiligo has also been achieved by transplantation of autologous melanocytes cultured on amniotic membrane, 241 or in fibrin suspension. 242 Repigmentation occurs in at least 70% of the treated area; it is caused primarily by the transplanted melanocytes. 243 A new commercial kit, Recell®, is a portable battery operated cell-harvesting device which is easy to use. 236 Further trials are needed. Autologous minigrafting (see above), and suction blister epidermal grafting are other surgical techniques.244. and 245. Plaques of verruca vulgaris have developed in grafted skin. 246
A recent Cochrane review concluded that the best evidence from individual trials showed ‘short-term benefit from topical steroids and various forms of UV light with topical preparations’. 247
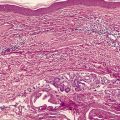
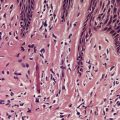
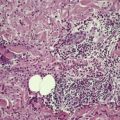
As a general rule, vitiliginous skin shows a complete loss of melanin pigment from the epidermis and an absence of melanocytes (Fig. 10.1). At the advancing border the melanocytes may be increased in size with an increased number of dendrites (Fig. 10.2). 44 Occasional lymphocytes may be present in this region; 248 these cells are invariably present if there is an inflammatory border clinically. 249 Sometimes these epidermotropic lymphocytes form small Pautrier-like collections in the basal layer. 250 In these instances there is also a perivascular infiltrate of mononuclear cells involving the superficial plexus, as well as some superficial edema. 251 A heavy lymphocytic infiltrate in the upper dermis is a rare finding. 252 In a recent study of 210 cases of vitiligo marginally active lesions with erythema, scaling, and hyperpigmentation were identified in 13% of cases. Lymphocytic infiltration of the dermoepidermal interface was observed in 89% of these cases. 253 Focal spongiosis is sometimes present in the marginal areas of vitiligo. 250 Ultrathin sections will often show vacuolated keratinocytes and extracellular granular material in the basal layer of the normal skin adjacent to areas of vitiligo. 254 If serial sections are examined, a lymphocyte will sometimes be found in close apposition to a melanocyte at the advancing edge (Fig. 10.3). 250 Degenerative changes have also been reported in nerves and sweat glands. 255 Merkel cells were absent from lesional skin in one study. 256 Langerhans cells are usually increased. 257 For this reason, it is the author’s practice to perform a melan-A and HMB-45 stain on all suspected cases of vitiligo; this allows the proper assessment of melanocyte numbers. Molecular studies have found that some lesions of vitiligo show focal melanocyte survival. 258 This has been confirmed by a histopathological study of 100 cases of vitiligo. In 12 of the cases some melanocytes were present. In 16% of cases there was some melanin in the basal layer with the Masson–Fontana stain. 259 Melanocytes are always reduced more in vitiligo than they are in nevus depigmentosus. 259
Vitiligo. (A) Melanocytes and melanin are absent from the basal layer. (H & E) (B) No melanin can be seen in the basal layer or dermis in this stain for melanin. (Masson–Fontana)
Vitiligo. A melanocyte with a giant melanosome is present at the edge of the depigmented area. (H & E)
Vitiligo. A lymphocyte is present next to a melanocyte showing early apoptosis. Melanocytes are absent elsewhere in the basal layer. (H & E)
The incidence of actinic damage and various skin cancers is surprisingly low in vitiligo patients, possibly because they practice sun-protection strategies. 260
Experimentally, if minor trauma is applied to non-lesional vitiligo skin, melanocytes become detached and undergo transepidermal elimination. 261 This may be the mechanism of the depigmentation occurring in the Koebner phenomenon. 261
Melanocytes are absent from lesions of long standing. 262 Melanocytes and keratinocytes adjacent to the vitiliginous areas show degenerative changes in the form of intracellular edema and vacuolar formation.254.262.263. and 264. Extracellular material derived from degenerating keratinocytes is sometimes present. 254 Fibrillar masses similar to colloid bodies may also be present in the upper dermis and in the basal layer. 254 Similar changes are seen in stable vitiligo, indicating ongoing damage to keratinocytes, melanocytes, and Langerhans cells resembling, in part, the changes seen in the lichenoid reaction. 265 Numerous nerve endings may be seen in close contact with the basal lamina. 263 There may be increased thickness of the basement membrane of Schwann cells and features of both axonal degeneration and nerve regeneration. 266
Oculocutaneous albinism is a genetically heterogeneous group of disorders in which there is a generalized decrease or absence of melanin pigment in the eyes, hair, and skin.267. and 268. At least 10 forms of this condition have been identified, each presumably resulting from a different biochemical block in the synthesis of melanin (Table 10.2). 6 The most common forms are types 1 and 2, which account for 40% and 50%, respectively, of cases worldwide. 269 Ocular albinism, in which the pigmentary deficit is confined to the eyes, will not be considered. 270
In type 1A, the classic type (OMIM 203100), the defect is a complete absence of tyrosinase activity in melanocytes. 271 The tyrosinase gene (TYR) has been cloned. It is present on chromosome 11q14–q21.272.273. and 274. Many different mutations of type 1A have been described.272.275. and 276. Prenatal diagnosis of type 1A can be made by performing a dopa test on the hair bulbs of fetuses, obtained by scalp biopsy. 277 This technique has been superseded by analysis of the fetal tyrosinase gene. 278 Inheritance is autosomal recessive in type. The clinical presentation at birth is white hair and skin and blue eyes.
Ocular disorders include photophobia, nystagmus, strabismus, and reduced visual acuity. In the skin there is accelerated photoaging and an increased incidence of keratoses and squamous and basal cell carcinomas.279. and 280. Malignant melanomas develop occasionally. 281 The dysplastic nevus syndrome has also been reported in individuals with oculocutaneous albinism. 282 Lentigines and nevi do not form in type 1A, the tyrosinase-negative phenotype. 6
In all phenotypes except type 1A, there is some increase in pigment with age, the amount depending on the ethnic background of the individual and the particular subtype of the disorder. 267 Red-yellow pheomelanin is the first to form; black-brown eumelanin is synthesized only after a long period of pheomelanin formation. 267
In yellow mutant oculocutaneous albinism (type 1B – OMIM 606952), tyrosinase activity and melanin biosynthesis are greatly reduced. 276 There is extreme hypopigmentation at birth with the eventual development of yellow or blond hair. A splicing mutation of the tyrosinase gene (TYR) on chromosome 11q14–q21 has been reported. 283
In oculocutaneous albinism type 2 (OMIM 203200), an autosomal recessive disease, there is defective melanin production in the skin, hair, and eyes. 284 It is caused by mutations of the P gene (OCA2), located on chromosome 15q11–q13, and thought to act as a transporter in the melanosomal membrane. 285 The P gene is deleted in the majority of patients with Angelman syndrome (OMIM 105830) and Prader–Willi syndrome (OMIM 176270). 284 This variant (OCA2) is common in some parts of Africa; in Tanzania its incidence is 1 in 1400 people per year. 286 Skin cancer is a problem in some of these individuals. 286
In tyrosinase-positive oculocutaneous albinism, one of the commonest genetic conditions in Africa, there is also a defect in chromosome 15q11–q13. It appears to be a phenotypic variant of type 2. Palmoplantar freckles and melanocytic nevi occur in a significant number of subjects with this form of the disease. 287
This variant (OMIM 203290) is caused by mutations in the tyrosinase-related protein-1 (TYRP1) gene located at 9p23. It is found predominantly in Africans although it has been reported in a consanguineous Pakistani family. 269 The so-called rufous variant (OMIM 278400), which occurs in black individuals, is characterized by bright copper-red coloration of the skin and hair and dilution of the color of the iris. 269 It is also due to a mutation in the TYRP1 gene.
The Hermansky–Pudlak syndrome (OMIM 203300) is a rare, autosomal recessive disorder of lysosome-related organelle biosynthesis resulting in melanosome dysfunction and absent platelet dense bodies. 291 Eight subtypes of Hermansky–Pudlak syndrome (HPS) have been identified (HPS-1 through HPS-8); all exhibit oculocutaneous albinism and absent platelet dense bodies.291. and 292. Additional features are present in each subtype, which may include pulmonary fibrosis and immunodeficiency. Several subtypes are limited to single case reports. Lipid and ceroid pigment are present in macrophages in various organs, including the skin. 293 Pulmonary ceroid deposition leading to respiratory failure is a common cause of death in HPS-1 and HPS-4.291. and 294. The HPS-1 gene, mutations of which are responsible for the Hermansky–Pudlak syndrome type 1, maps to chromosome 10q23.295. and 296. Mutations cause lysosomal dysfunction in platelets and melanocytes, possibly by affecting calcium channel integrity in the cells.297. and 298. Other cutaneous findings, most often related to a specific 16-base pair duplication of the HPS1 gene, include dysplastic nevi, acanthosis nigricans-like lesions in the neck and axilla, and trichomegaly. 295 There is a suggestion that a patient with HPS-1 may be predisposed to the development of systemic lupus erythematosus. 299
Hermansky–Pudlak syndrome type 2 (OMIM 608233), the only other subtype that will be considered here, is caused by a mutation in the gene encoding the β-3A subunit of the AP3 complex (AP3B1). It includes immunodeficiency in its phenotype, and patients have an increased susceptibility to infections as a consequence of neutropenia. The gene maps to 5q14.1.
The Chédiak–Higashi syndrome, the Griscelli syndrome, and the Elejalde syndrome (see below) are sometimes regarded as other clinical variants of oculocutaneous albinism. They have in common the presence of silvery hair and mild skin coloration which is not strictly albinism.
Generalized cutaneous depigmentation resembling albinism has been reported in a patient treated with a sulfonamide. However, the absence of melanocytes on electron microscopy was more in keeping with vitiligo or chemical leukoderma. 300
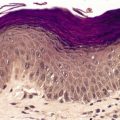
There is a complete or partial reduction in melanin pigment in the skin and hair bulbs. Melanocytes are normal in number and morphology (Fig. 10.4). Tyrosinase activity is lacking in melanocytes in freshly plucked anagen hair bulbs in type 1A; 301 it is reduced in heterozygotes with this phenotype and variably reduced in some of the other types. Tyrosinase activity is normal in type 2. 271
Albinism. Melanin is absent from the basal layer but melanocytes are normal in number and morphology. (H & E)
The Chédiak–Higashi syndrome (OMIM 214500) is a rare, autosomal recessive disorder in which there is partial oculocutaneous albinism associated with frequent pyogenic infections and the presence of abnormal, large granules in leukocytes and some other cells.304.305.306. and 307. The Hermansky–Pudlak syndrome (see above) is a similar, but distinct entity. The disease usually enters an accelerated phase in childhood, with pancytopenia, hepatosplenomegaly, and lymphohistiocytic infiltrates in various organs. 308 This phase, which resembles the virus-associated hemophagocytic syndrome, is usually followed by death. 308
The pigmentary dilution involves at least one and often all three of the following – skin, hair, and eyes.304. and 309. There is increased susceptibility to burning. The hair is usually blond or light brown in color. Speckled hypopigmentation and hyperpigmentation of sun-exposed areas is sometimes found in darkly pigmented races. 310
The increased susceptibility to infection is related to impaired function of leukocytes and natural killer cells associated with lysosomal defects, while the reduced skin pigmentation is related to similar defects in melanocytes.308. and 311. The inclusions found in these and other cells are massive secondary lysosomal structures formed through a combined process of fusion, cytoplasmic injury, and phagocytosis.308. and 311.
The gene responsible for this condition maps to chromosome 1q42.1–q42.2. The gene has been designated LYST (lysosomal trafficking regulator).
There is a striking reduction or even absence of melanin pigment in the basal layer and in hair follicles. 304 A few large pigment granules corresponding to giant melanosomes are present. 312 In less affected individuals and in some heterozygotes, clumps of enlarged pigment granules may also be present in the dermis in macrophages and endothelial cells and lying free in the interstitium. 309
Staining with toluidine blue demonstrates large cytoplasmic inclusions in cutaneous mast cells. 312
Giant melanosomes and degenerating cytoplasmic residues are found in melanocytes. 313 The pigment granules passed to keratinocytes are bigger than normal. 313 The giant melanosomes appear to arise from defective premelanosomes. 313 Giant cytoplasmic granules have also been found in Langerhans cells. 314 They are believed to be derived from the fusion of lysosomes or some portion of Birbeck granules. 314
The Griscelli syndrome is characterized by reduced skin pigmentation, often regarded as partial albinism, and silvery-gray hair combined in one type with immunodeficiency.315. and 316. Three types have been described: type 1 (OMIM 214450) caused by mutations in the myosin VA gene (MYO5A) at chromosome 15q21, and without immunological impairment; type 2 (OMIM 607624) caused by mutations in the RAB27A gene at 15q21, the same location as the MYO5A gene, and with immunological impairment; 317 and type 3 (OMIM 609227) characterized by hypomelanosis with no immunological or neurological changes and caused by mutations in the melanophilin (MLPH) gene at 2q37, or the MYO5A gene. 318 There are no abnormal cytoplasmic granules in leukocytes as found in the Chédiak–Higashi syndrome (see above).
Enlarged hyperpigmented basal melanocytes with sparsely pigmented adjacent keratinocytes are seen on skin biopsy specimens. 319
There are some type IV melanosomes in basal melanocytes and shortened dendritic processes. The hair shafts show uneven clusters of aggregated melanin pigment, mainly in the medulla. 315
The Elejalde syndrome (OMIM 256710) is a rare autosomal recessive disorder characterized by the triad of silvery hair, hypopigmented skin (sometimes referred to as partial albinism), and severe dysfunction of the central nervous system (hypotonia, seizures, and mental retardation). 320 It has also been called neuroectodermal melanolysosomal disease. 321 There is no immunodeficiency as seen in the type 2 Griscelli syndrome or Chédiak–Higashi syndrome (see above), which share clinical features with Elejalde syndrome. It has been proposed that Elejalde syndrome and Griscelli syndrome type 1 (OMIM 214450) may represent the same entity. 322 If so, it is caused by a mutation in the gene encoding myosin VA (MYO5A), which maps to chromosome 15q21.
Melanin granules in the basal layer are of irregular size and distribution with overall reduced pigmentation. Hair shafts are similar to those seen in the Griscelli syndrome (see above).
Progressive macular hypomelanosis of the trunk is an acquired form of hypopigmentation with a predisposition to affect the back of young adult females of Caribbean origin. 323 It has also been reported from other countries, including Singapore. 324 The hypopigmented macules, which measure 1–3 cm in diameter, coalesce into large patches. The disease is often misdiagnosed as tinea (pityriasis) versicolor. 325 The disease may remit after 3–4 years. A recent study has suggested that idiopathic guttate hypomelanosis (see below) is a related disorder along a spectrum of disorders of depigmentation. 326 Extensive pityriasis alba was thought be the same condition, 327 but this theory has since been rejected. 328 On the basis of red follicular fluorescence in hypopigmented spots, and culture results, it has been proposed that progressive macular hypomelanosis may result from Propionibacterium acnes.329. and 330.
The disease does not respond to conventional treatment including PUVA, but a case has been successfully treated with narrowband UVB. 331 In a recent randomized study antimicrobial therapy in conjunction with light (benzoyl peroxide/clindamycin/UVA) was more effective than a combination of anti-inflammatory therapy and light (fluticasone/UVA). 330 Improvement occurred in a recently reported case with sunlight exposure and doxycycline. 325
There is a decrease in melanin pigment within the epidermis. Melanocytes are normal in number.
There is a reduction in stage IV (negroid) melanosomes, which are replaced by small type I–III melanosomes in an aggregated (caucasoid) pattern. This results in a decrease of epidermal melanin. 332
Tuberous sclerosis (OMIM 191100) is characterized by the triad of epilepsy, mental retardation, and multiple angiofibromas (‘adenoma sebaceum’) (see p. 810). In addition, circumscribed macules of hypopigmentation known as ‘ash leaf spots’ can be present at birth on the trunk and lower extremities.6. and 333. They vary in diameter from 1 mm to 12 cm. The more common shapes are oval, polygonal, or ash leaf-like. The basic abnormality appears to be an arrest in the maturation of melanosomes. 6 Tuberous sclerosis exhibits genetic heterogeneity. This entity is discussed further in Chapter 34, page 810.
Epidermal melanin is reduced, but not absent.
Electron microscopy has shown a normal number of melanocytes and a reduction in the number, size, and melanization of the melanosomes. 333 The small melanosomes often form aggregates within the keratinocytes.
Idiopathic guttate hypomelanosis is a common leukodermic dermatosis of unknown etiology in which multiple achromic or hypochromic macules, 2–5 mm in diameter, develop over many years.335.336.337. and 338. They are usually found on the sun-exposed extremities of elderly individuals, but scattered lesions may occur on the trunk.339. and 340. Repigmentation does not occur. The author and colleagues have seen this pattern of pigmentation in patients who have received bone marrow transplants (unpublished observation). In a group of renal transplant patients, the presence of this condition correlated with the presence of HLA-DQ3. 341
There is a decrease in melanin pigment in the basal layer of the epidermis and a reduction in the number of dopa-positive melanocytes, although these cells are never completely absent. The epidermis usually shows some atrophy, with flattening of the rete pegs. There may be basket-weave hyperkeratosis.
Hypomelanosis of Ito (incontinentia pigmenti achromians – OMIM 300337) presents at birth or in infancy with sharply demarcated, hypopigmented macular lesions on the trunk and extremities, with a distinctive linear or whorled pattern distributed along the lines of Bla-schko.345.346.347.348.349. and 350. The pattern resembles a negative image of the pigmentation seen in incontinentia pigmenti (see p. 297). 351 The coexistence of hypomelanosis of Ito and incontinentia pigmenti in the same family, even though disputed by a subsequent author, 352 and the report of several patients with a preceding erythematous or verrucous stage353. and 354. have led several authorities to postulate a link between these two conditions.346. and 353. This seems unlikely in our current state of knowledge. The coexistence of hypomelanosis of Ito and whorled hypermelanosis has been reported. 355
Other features of hypomelanosis of Ito include a female preponderance, a tendency for lesions to become somewhat pigmented in late childhood, a family history in a few cases, 356 and the coexistence in a high percentage of patients of abnormalities of the central nervous system (particularly seizures and mental retardation), eyes, hair, teeth, and musculoskeletal system.345.346.357.358. and 359. Ileal atresia and leptomeningeal angiomatosis are other associated abnormalities.360. and 361. In one patient, a streptococcal exanthem developed in a Blaschko-linear pattern. 362
Many different chromosomal abnormalities have been recorded in this condition, leading to a suggestion that this is not a discrete diagnostic condition but is rather a symptom of many different states of mosaicism.348. and 358. The term pigmentary mosaicism has been suggested as a better title. 363 Incontinentia pigmenti type 1, which was subsequently shown to be hypomelanosis of Ito, is associated with an X autosome translocation involving Xp11. Other chromosomal abnormalities have included trisomy 2 mosaicism, 364 and trisomy mosaicism for chromosomes 7, 12, 13, 14, 15, and 18. 364 Ring chromosome 10, and supernumerary X-chromosome ring fragments have also been described. 364 It has also been reported in mosaic Turner’s syndrome. 365
The hypopigmented areas show a reduction in melanin pigment in the basal layer, but this is usually not discernible in hematoxylin and eosin-stained sections and requires a Masson–Fontana stain for confirmation. Dopa stains show a reduction in staining of melanocytes and sometimes shortening of their dendrites. 366 A reduction in the number of melanocytes367 and vacuolization of basal keratinocytes have been mentioned in some reports but specifically excluded in most. 368
Electron microscopy has shown a reduction in melanosomes in melanocytes in the hypopigmented areas and a decrease in the number of melanin granules in keratinocytes. 345 There are isolated reports of aggregation of melanosomes, vacuolization of melanocytes, 263 and an increase in the number of Langerhans cells in the epidermis. 369
Nevus depigmentosus (achromic nevus) is a rare entity consisting of isolated, circular or rectangular, hypopigmented macules with a predisposition for the trunk and proximal parts of the extremities.6. and 370. In one study, nearly one-half had only one lesion, but a fifth of patients had more than 10 lesions. 371 It may also occur along Blaschko’s lines or in a systematized pattern, the latter having some clinical resemblance to the pattern seen in hypomelanosis of Ito.334. and 372. In the majority of cases the lesions are present at birth or appear in early childhood. 373 Lesions remain stable over time. 371 Under Wood’s lamp, lesions have an off-white accentuation without fluorescence. 370 Systemic lesions are uncommon374 but an association with unilateral lentiginosis and ILVEN (see p. 669) has been reported.375. and 376. Ipsilateral involvement of the iris has been reported. 377 Two sisters who presented with guttate and nummular hypomelanosis in a segmental distribution had numerous melanocytes on biopsy but the melanocytes had a decreased number of melanosomes. 378
One study found a selective defect in eumelanogenesis in nevus depigmentosus, although this remains to be confirmed. 379
There are several reports of autologous epidermal grafts being used to treat this condition. They have had mixed success.380. and 381. In another case, misdiagnosed clinically as segmental vitiligo, and treated with prolonged intense UVB therapy, lentigines developed in the achromic lesion. 382
The melanin content of lesional skin is decreased compared with perilesional normal skin. 370 Melanocytes are said to be normal or slightly reduced in number, although there is reduced dopa activity.6. and 373. In one study, melanocyte counts were significantly reduced with MART-1 and GP-100 stains for melanocytes. 371 In a recent study of 30 cases, the number of melanocytes was decreased, but not as much as in vitiligo. 259
Melanosomes are normal in size but there may be abnormal aggregation of them within melanocytes. 379 One study showed a reduction of melanosomes in melanocytes. 373 Degradation of melanosomes within autophagosomes of melanocytes has been noted. Melanosomes are decreased in number in keratinocytes, suggesting impaired transfer. 374
Pityriasis alba consists of variably hypopigmented, slightly scaly patches, varying from 0.5 to 6 cm in diameter with a predilection for the face, neck, and shoulders of dark-skinned atopic individuals.383.384. and 385. It is more common in males than females, and most patients are between 6 and 16 years of age. 386 The etiology is unknown although it has been regarded as postinflammatory hypopigmentation following eczema. 383 It has been reported in patients with atopic eczema. 387 The role of organisms is controversial; none has been confirmed as a causal factor. 386
A supposed variant with extensive non-scaling macules involving the lower part of the trunk has been reported, but there is no real evidence that this is the same process.384. and 385.
Pityriasis alba is a frequent reason for medical consultation because of its chronic course, tendency to relapse, and esthetic impact. 388 Topical corticosteroids are often used in the treatment of this condition even though no formal study has assessed their efficacy. 388 However, long-term use of topical corticosteroids is not desirable, particularly on the face. Recently, tacrolimus ointment (0.5%) was reported to produce satisfactory results. 388 It should be noted that tacrolimus is not approved in some countries for pediatric use.
Another suggested clinical variant is pigmenting pityriasis alba, in which a central zone of bluish hyperpigmentation develops in a scaly, hypopigmented patch. 389 A dermatophyte was present in 65% of these cases. 389
There are no detailed studies of the usual facial type of pityriasis alba. In a personally studied case there was mild hyperkeratosis, focal parakeratosis and focal mild spongiosis with prominent exocytosis of lymphocytes. 390 There was also a mild superficial perivascular inflammatory cell infiltrate in the dermis. Melanin pigmentation of the basal layer was markedly reduced, but there was no melanin incontinence. 390 Melanocytes were normal in number. This conforms with one other reported case. 391 A reduced number of melanocytes with smaller melanosomes is another suggested finding. 389 Melanocytes were normal in number in a recent study of 56 cases. 392
A study of the ‘extensive’ variant showed reduced basal pigmentation, a decreased number of functional melanocytes on the dopa preparation, and a reduction in the number and size of melanosomes. 384
Hypopigmented areas may develop during the course of a number of inflammatory diseases of the skin, usually during the resolving phases. 6 Examples include the various eczematous dermatitides, psoriasis, discoid lupus erythematosus, pityriasis rosea, variants of parapsoriasis, lichen sclerosus et atrophicus, syphilis and the viral exanthems. 6 Uncommonly, hypopigmentation may follow lichen planus and other lichenoid eruptions. It may occur in the vicinity of the injection site, following the injection of corticosteroids; 394 it may follow the application of imiquimod cream. 395 Extensive hypopigmentation has followed the commencement of antiretroviral treatment in an HIV-seropositive African woman. 396 Hypomelanotic lesions may occur at an early stage, albeit uncommonly, in some of the following diseases: alopecia mucinosa, sarcoidosis, mycosis fungoides, pityriasis lichenoides chronica, pityriasis versicolor (tinea versicolor), onchocerciasis, yaws, and leprosy. 6
The mechanism of the hypopigmentation in many of these conditions is thought to be a block in the transfer of melanosomes from melanocytes to keratinocytes; in the lichenoid dermatoses damage to melanocytes may also contribute. In pityriasis versicolor, melanosomes are poorly melanized; impaired transfer is also present.
Various mechanisms have been proposed for the hypopigmentation of lesions in indeterminate and tuberculoid leprosy (see p. 564).
There is a reduction in melanin pigment in the basal layer, although not a complete absence. Melanocytes are usually normal in number. Pigment-containing melanophages are sometimes present in the upper dermis, particularly in black patients. Residual features of the preceding or concurrent inflammatory dermatosis may also be present.
Nevus anemicus is an uncommon congenital disorder in which there is usually a solitary asymptomatic patch that is paler than the surrounding normal skin. 401 Its margin is irregular and there may be islands of sparing within the lesion. 402 The pale area averages 5–10 cm in diameter. There is a female predominance. 403 There is a predilection for the upper trunk, although involvement of the face, neck, and extremities occurs.403. and 404. A variant with multiple lesions on the arms has been reported. 405 Nevus anemicus sometimes occurs in association with neurofibromatosis, 406 phakomatosis pigmentovascularis, or port wine stains. 407 Onset in one patient was at age 21 years, suggesting the likelihood of an acquired variant of nevus anemicus. 403Bier’s spots have a similar appearance, but they are permanent, often associated with venous stasis. They probably result from anatomical or functional damage to small blood vessels.408. and 409.
Nevus anemicus is regarded as a pharmacological nevus in which the pallor is attributable to increased sensitivity of the blood vessels in the area to catecholamines. 410 It has been found that the vessels do not respond normally to proinflammatory cytokines, at least at the level of E-selectin expression. 411Nevus oligemicus is a related entity in which there is livid erythema rather than pallor.412. and 413.
No abnormalities have been shown by light or electron microscopy.
The disorders characterized by hyperpigmentation constitute a heterogeneous group of diseases comprising a bewildering number of rare conditions. Japanese people are predisposed to many of the entities to be discussed below. Several factors are taken into consideration in the clinical categorization of these various disorders, including the distribution, arrangement, and morphology of individual lesions as well as the presence or absence of hypopigmented areas.414. and 415. Four clinical categories of hyperpigmentation can be recognized.
1. Diffuse hyperpigmentation: generalized hyperpigmentary disorders (scleroderma, Addison’s disease, myxedema, Graves’ disease, malnutrition including pellagra, chronic liver disease including hemochromatosis and Wilson’s disease, porphyria, folate and vitamin B12 deficiency, heavy metal toxicity and the ingestion of certain drugs and chemicals), universal acquired melanosis, the generalized melanosis that may develop in malignant melanoma, and in some cases of pheochromocytoma. 416
2. Localized (patchy) hyperpigmentation: ephelis (freckle), café-au-lait spots, macules of Albright’s syndrome, macules of Peutz–Jeghers syndrome, macules of Laugier–Hunziker syndrome, Becker’s nevus, acromelanosis, melasma, fixed drug eruption, frictional melanosis, notalgia paresthetica, familial progressive hyperpigmentation and idiopathic eruptive macular pigmentation; the boys who presented with pigmented hypertrichotic lesions on the upper inner thighs with variable involvement of the genitalia, trunk, and limbs, associated with insulin-dependent diabetes are difficult to classify. 417
3. Punctate, reticulate hyperpigmentation (including whorls and streaks): Dowling–Degos disease, Kitamura’s disease, Naegeli–Franceschetti–Jadassohn syndrome, dermatopathia pigmentosa reticularis, macular amyloidosis, ‘ripple neck’ in atopic dermatitis, hereditary diffuse hyperpigmentation, incontinentia pigmenti, prurigo pigmentosa, confluent and reticulated papillomatosis, patterned hypermelanosis, and chimerism; generalized mottled pigmentation with acral blistering can also be grouped here. 418
4. Dyschromatosis (hyperpigmentation and hypopigmentation): dyskeratosis congenita, dyschromatosis symmetrica hereditaria (Dohi), dyschromatosis universalis hereditaria, familial gigantic melanocytosis, heterochromia extremitarum, 415 Fanconi anemia, 419 and hereditary congenital hypopigmented and hyperpigmented macules. 420
Theoretically, the hyperpigmentation observed in these various conditions could result from increased basal pigmentation and/or melanin incontinence. Alterations in the epidermal configuration can also produce apparent pigmentation of the skin.
Although there is some variability in the histopathological features reported in some of the disorders of hyperpigmentation, the following subclassification provides a useful approach to a biopsy from such a disease:
• Disorders with basal hyperpigmentation (mild melanin incontinence is sometimes present also): generalized hyperpigmentary disorders, universal acquired melanosis, acromelanosis (increased melanocytes were noted in one report), familial progressive hyperpigmentation, idiopathic eruptive macular pigmentation, dyschromatosis symmetrica hereditaria, dyschromatosis universalis, patterned hypermelanosis, chimerism, melasma, acquired brachial dyschromatosis, ephelis (freckle), café-au-lait spots, macules of Albright’s syndrome, Laugier–Hunziker syndrome, Bannayan–Riley–Ruvalcaba syndrome and Peutz–Jeghers syndrome, and Becker’s nevus (melanosis).
• Disorders with epidermal changes: Dowling–Degos disease (the epidermal changes resemble those of solar lentigo), Kitamura’s disease (the epidermal changes resembling Dowling–Degos disease but with intervening epidermal atrophy also), and confluent and reticulated papillomatosis of Gougerot–Carteaud (the epidermal changes are those of papillomatosis).
• Disorders with striking melanin incontinence: postinflammatory melanosis, prurigo pigmentosa, generalized melanosis in malignant melanoma, dermatopathia pigmentosa reticularis, Naegeli–Franceschetti–Jadassohn syndrome, incontinentia pigmenti, and late fixed drug eruptions.
• Disorders with melanin incontinence and epidermal atrophy or ‘dyskeratotic’ cells: dyskeratosis congenita, frictional melanosis, notalgia paresthetica, ‘ripple neck’ in atopic dermatitis, active fixed drug eruptions, and active prurigo pigmentosa.
Fixed drug eruptions and dyskeratosis congenita are discussed with the lichenoid reaction pattern on pages 50 and 68 respectively. Confluent and reticulated papillomatosis is considered on page 505.
As mentioned above, generalized cutaneous hyperpigmentation can be seen in a number of metabolic, endocrine, 421 hepatic, and nutritional disorders, as well as following the application of topical calcipotriene (calcipotriol), 422 and the intake of certain drugs423 and heavy metals. 424 Hyperpigmentation may follow sympathectomy. 425 It may also occur in the POEMS (Crow–Fukase) syndrome426 (see p. 900).
Biopsies of the pigmented skin are not often taken from individuals with these conditions. There is an increase in melanin in the lower layers of the epidermis and sometimes a small amount of pigment in the dermis. Of interest is the finding of large nuclei in the keratinocytes of the pigmented skin in some megaloblastic anemias.427. and 428.
Hemosiderin pigment was present around dermal capillaries and sweat glands in two cases of hyperpigmentation associated with hyperthyroidism. 429
Universal acquired melanosis is an extremely rare condition, also known as the ‘carbon baby’ syndrome. It is characterized by progressive pigmentation of the skin during childhood, resembling that seen in black races. 430
In the reported case there was hyperpigmentation of the epidermis and an increase in type III and IV (negroid pattern) melanosomes in melanocytes. 430
Acromelanosis refers to the presence of pigmented patches and macules on the dorsal surface of the phalanges, usually in colored people.414. and 415. Several clinical variants have been recognized on the basis of the distribution of the pigment and the progression of the disorder.415. and 431. Hyperpigmentation of the distal phalanges of both hands and feet is usually a prominent feature of dark-skinned newborns, as is hyperpigmentation of the external genitalia and areola. 432 Periungual hyperpigmentation has also been reported in a small number of fair-skinned newborns. 432 It fades with time.
Basal hyperpigmentation is the usual finding, although an increase in basal melanocytes with associated acanthosis has also been reported. 431
Patches of hyperpigmentation are present at birth in this rare genodermatosis. 433 They increase in size and number with age. Eventually a large percentage of the skin and mucous membranes becomes hyperpigmented. 433
The most striking change is an increase in melanin pigment within the epidermis, especially in the basal layer. There is some concentration at the tips of the rete ridges.
Idiopathic eruptive macular pigmentation is an exceedingly rare condition characterized by asymptomatic, pigmented macules involving the neck, trunk, and proximal extremities.434. and 435. Fewer than 30 cases have been reported. 436 This idiopathic disorder involves children and adolescents; one case was associated with pregnancy and Hashimoto thyroiditis. 436 The lesions usually appear abruptly. Spontaneous resolution can be expected within several months to a few years,434. and 437. although a case lasting 21 years, with several episodes of spontaneous resolution followed by recurrences, has been reported. 438
There is increased pigmentation of the basal layer, pigmentary incontinence with many melanophages in the upper dermis, and a sparse perivascular lymphohistiocytic infiltrate. 439 The changes are similar to those seen in erythema dyschromicum perstans, to which this disorder has been likened; 439 others disagree. 435
Dyschromatosis symmetrica hereditaria (OMIM 127400), also known as reticulate acropigmentation of Dohi, 440 consists of freckle-like lesions on the dorsum of the hands and feet with scattered depigmented macules in between.414.415.441.442. and 443. The age of onset is approximately 6 years. Cases from Japan, China, and Korea generally have an autosomal dominant pattern of inheritance. This form of the disease results from mutations in the double-stranded RNA-specific adenosine deaminase (ADAR/DSRAD) gene located at 1q11–q21.444.445.446.447. and 448. Another study mapped this gene to 1q21–q22, 449 while the OMIM gene map lists it at 1q21.3. Another study lists the gene location as 1q11–q12. 450 The gene encodes the enzyme responsible for the deamination of adenosine to inosine; 445 as such it is an RNA editing enzyme. 451 Several cases reported recently from the Middle East had autosomal recessive inheritance. 452 Nothing is known about the genetic abnormality in these cases.
Both dyschromatosis symmetrica hereditaria and dyschromatosis universalis hereditaria (see below) are inherited pigmentary skin disorders. In the latter condition skin lesions occur earlier (first month of life) and truncal involvement is usually present, while the lesions in dyschromatosis symmetrica hereditaria are predominantly acral in location. 445
The epidermis shows increased pigmentation, mainly basal, in the hyperpigmented areas, and reduced pigmentation, sometimes accompanied by a reduction in the number of melanocytes, in hypopigmented areas. 415
Dyschromatosis universalis hereditaria (OMIM 127500) is the prototype condition for a group of dyschromatoses characterized by areas of hypopigmentation and hyperpigmentation.414.453.454. and 455. The absence of atrophy and telangiectasia distinguishes this group from the poikilodermas. 414 Onset is in early childhood, usually the first few months of life, with involvement being most prominent on the trunk and extremities. 445 This contrasts with dyschromatosis symmetrica hereditaria (see above) in which lesions are almost always acral in location. Clinical variants have been described.456.457.458.459. and 460.
The gene responsible maps to chromosome 6q24.2–q25.2. 445 While most cases have had an autosomal dominant inheritance, an autosomal recessive inheritance seems likely in some cases. 461 In the autosomal recessive variant, a new locus on 12q21–q23 appears to be involved. 462
There is variable epidermal pigmentation which may be accompanied by some pigment incontinence. The number of melanocytes is sometimes reduced in the hypopigmented areas. 456
The term ‘patterned hypermelanosis’ is proposed for several rare dermatoses with overlapping features which have been reported in the past by different names. They are characterized by linear, whorled, or reticulate areas of hyperpigmentation. 415 Although the term ‘zosteriform’ has been used to describe the pattern of the pigmentation in some of these cases, it has been pointed out that this term has not always been used correctly; the hyperpigmentation usually follows Blaschko’s lines (the boundary lines separating areas of the skin subserved by different peripheral nerves) and not the courses of the nerves themselves as in a zosteriform pattern.463.464.465. and 466. A review of 54 children with segmental, linear, or swirled hyper- and/or hypopigmentation along the lines of Blaschko revealed that 16 had extracutaneous manifestations. 350 Another study in which pigmentary anomalies along the lines of Blaschko were associated with abnormalities of the central nervous system included a few patients with incontinentia pigmenti and hypomelanosis of Ito; most cases could not be categorized further. 467 In another report, hyperpigmentation along the lines of Blaschko was associated with chromosome 14 mosaicism. 468 Dyspigmentation has also been associated with mosaic chromosome 5p tetrasomy469 and with trisomy 20 mosaicism. 470
Included in the patterned hypermelanoses are cases reported as ‘linear and whorled nevoid hypermelanosis’,463.465.471.472.473.474.475.476.477.478. and 479. ‘reticulate hyperpigmentation distributed in a zosteriform fashion’, 480‘progressive cribriform and zosteriform hyperpigmentation’,481. and 482. ‘congenital curvilinear palpable hyperpigmentation’, 483‘zebra-like hyperpigmentation’, 484‘progressive zosteriform macular pigmented lesions’, 485‘dyschromia in confetti’ (following topical immunotherapy with diphenylcyclopropenone), 486 and ‘infant with abnormal pigmentation’. 487 A mottled pigmentation is seen in mosaic subclinical melanoderma, a condition observed when photo-exposed skin of adults is examined by ultraviolet light. 488 The term ‘patterned hypermelanosis’ is not applicable to well-defined entities such as incontinentia pigmenti and the reticulate acral pigmentations of Kitamura and of Dowling and Degos (see p. 295). Streaks of hyper- and hypopigmentation can be seen in the Pallister–Killian (Killian–Teschler–Nicola) syndrome (OMIM 601803) associated with tetrasomy of chromosome 12p.489.490. and 491.
This exceedingly rare condition, which may have autosomal dominant inheritance, was originally described as ‘familial melanopathy with gigantic melanocytes’. 492 It is characterized by a diffuse brown hyperpigmentation, admixed with raindrop hypopigmentation, which affects mainly exposed areas but also to a lesser extent unexposed areas.492. and 493. The pigmentary changes are accompanied by sparse axillary and pubic hair, and light-colored scalp and body hair.
The cause of the disorder is unknown but melanocytes in both hyper- and hypopigmented skin seem to be unable to deliver melanin to the surrounding keratinocytes. 492
The key histological feature is the presence of abnormally large melanocytes, at least three times the size of normal melanocytes. They are present in both hyper- and hypopigmented zones but there are fewer melanocytes in the hypopigmented areas. There are areas with hyperpigmented basal cells alternating with poorly pigmented skin.
Chimerism results from double fertilization of an ovum, producing an individual (a chimera) with differing sets of chromosomes. 494 Abnormalities of skin pigmentation, usually in the form of irregular areas of hyperpigmentation, are a rare manifestation of the chimeric state.494. and 495.
Melanin is increased in the basal layers of the epidermis in the hyperpigmented lesions. 494
Melasma (chloasma) is an acquired, chronic, recurrent symmetrical hyperpigmentation of the forehead and cheeks which develops in some women, especially those living in areas of intense UV radiation, who are pregnant or taking oral contraceptives.5.496.497.498. and 499. Its incidence in pregnancy varies from 15 to 50% or more; 500 this figure varies with ethnicity. It has also been reported in women taking isotretinoin501 or hormone replacement therapy; 502 the forearms are sometimes involved in this latter group.503. and 504. The hormonal basis for melasma is poorly understood. There is increased expression of α-melanocyte-stimulating hormone in lesional skin, 505 and increased expression of stem cell factor in the dermis and of its receptor c-kit in the epidermis. 506
There have been very few well-conducted trials on the treatment of melasma and this makes the comparison of outcomes difficult. 499 A review of various clinical trials was published in 2006. 507 It stated that the use of a broad-spectrum sunscreen is important, as is topical hydroquinone, the most common treatment for melasma. Other lightening agents include retinoic acid (tretinoin) and azelaic acid. 507 A recent randomized controlled trial found that a triple combination of fluocinolone acetonide, hydroquinone, and tretinoin was more effective than hydroquinone alone in Asian patients with moderate to severe melasma. 508 Chemical peels, 509 laser treatment, and intense pulsed light therapy are additional therapeutic modalities. 507 Recently, a trial of topical rucinol serum produced skin lightening after 3 months of use. 510 Rucinol inhibits the activity of both tyrosinase and tyrosinase-related protein-1 (TRP-1). 510
There is increased melanin in the epidermis, particularly in the basal layers. Melanin pigment is located in a ‘cap’ overlying the keratinocyte nuclei. 511 One study showed that melanocytes were increased in most cases, but that some cases showed a normal or even a decreased number. 511 Another study found normal melanocyte numbers in all cases. 512 Melanosomes are increased in basal and suprabasal keratinocytes. 511 Melanocytes have increased numbers of dendrites. 512 Mild pigment incontinence is sometimes present. Solar elastosis is more prominent than in normal skin. 513 Mast cells are also increased in number. 513
Acquired brachial (cutaneous) dyschromatosis was applied recently to the asymptomatic, gray-brown patches of pigmentation, occasionally interspersed with hypopigmented macules, found predominantly on the dorsum of the forearms, mostly bilateral, of middle-aged patients. 514 There was a predilection for women, many of whom had been taking antihypertensive drugs, especially angiotensin-converting enzyme (ACE) inhibitors. 514 Many patients also had Civatte’s poikiloderma of the neck.
The pigmented lesions showed epidermal atrophy, increased basal layer pigmentation, superficial telangiectases, and actinic elastosis. There was no pigmentary incontinence or amyloid.
The hypopigmented macules showed a decrease in pigmentation of the basal layer.
Ephelides (freckles) are small, well-defined, pigmented macules 1–2 mm in diameter with a predilection for the face, arms, and shoulder regions of fair-skinned individuals. They appear at an early age and may follow an episode of severe sunburn.
The epidermis appears normal in structure. The basal cells in the affected areas are more heavily pigmented with melanin than those in the surrounding skin and there is usually sharp delimitation of the abnormal areas from the normal (Fig. 10.5). There are normal numbers of melanocytes. 515
Freckle (ephelis). Melanin is increased in the basal layers of the epidermis but melanocytes are normal in number and morphology. (H & E)
Café-au-lait spots are uniformly pigmented, tan to dark brown macules which vary in size from small, freckle-like lesions to large patches 20 cm or more in diameter. 516 They may be present at birth or develop within the first few years of life. 517 Some cases appear to develop early in embryogenesis, possibly as early as 3 to 4 weeks of gestation. 518 They are found in approximately 15% of individuals.519.520. and 521. They are not increased in patients with tuberous sclerosis, contrary to common belief. 522 Multiple café-au-lait spots are a feature of neurofibromatosis;517.521. and 523. axillary freckling is often also present in these cases (see p. 874). Café-au-lait spots have also been reported in Bloom’s syndrome, Cowden’s disease, Fanconi’s anemia, ring chromosome syndromes, and ataxia-telangiectasia.521. and 524. Patients with Fanconi’s anemia (OMIM 227650), an autosomal recessive disorder due to a mutation in the FANCC gene, may also have macules of ‘guttate’ hypopigmentation. 419 A large achromic patch has also been reported in the Silver–Russell syndrome (OMIM 180860) in which cutaneous dyschromia, usually in the form of café-au-lait macules, is combined with musculoskeletal abnormalities, genitourinary malformations, and craniofacial dysmorphy. 525 Familial, multiple café-au-lait spots have also been reported without any evidence of coexisting disease. 526 They have been reported on the upper and lower eyelids in one patient in a pattern analogous to the ‘kissing’ nevus. 527
In hematoxylin and eosin preparations, the lesions resemble freckles, with basal hyperpigmentation but no apparent increase in the number of melanocytes. However, quantitative studies have shown a slight increase in melanocytes, which are accommodated in focally elongated rete ridges.528. and 529. This was confirmed in one ultrastructural study. 530 Giant melanin granules (macromelanosomes), measuring up to 6 µm in diameter and recognizable on light microscopy, can be seen in café-au-lait spots in many patients with neurofibromatosis. 531 The diagnostic significance of macromelanosomes is diminished by their absence in some children with neurofibromatosis and their presence in normal skin and other pigmented macular lesions.532.533.534. and 535.
Albright’s syndrome (McCune–Albright syndrome – OMIM 174800) is characterized by the triad of polyostotic fibrous dysplasia, sexual precocity, especially in the female, and pigmented macules. 536 These macules are large, often unilateral, and related to the side of the bone lesions. The outline of the macules is very irregular, in contrast to that of café-au-lait spots. The macules may follow Blaschko’s lines. 537
The syndrome is associated with a postzygotic somatic mutation of the GNAS1 gene that encodes the α subunit of the Gs protein. The gene map locus is 20q13.2. 538 This condition shows genomic imprinting, a process whereby genetic alleles responsible for a phenotype are derived from one parent only. 539
The lesions resemble freckles, showing hyperpigmentation of the basal layer. Rarely, macromelanosomes can be identified. 533
The Laugier–Hunziker syndrome is characterized by melanotic pigmentation of the mouth and lips which is frequently accompanied by longitudinal melanonychia.5.540.541.542. and 543. In a small number of cases, there are dark palmoplantar and interdigital lesions. Pigmented macules may also develop about the nails. Conjunctival, vulval, and penile pigmentation occur rarely.544. and 545. There are usually no associated internal disorders. The occurrence of a hypocellular marrow in one patient may have been a fortuitous association. 546 Addison’s disease developed in another patient with features suggestive of Laugier–Hunziker syndrome. 547 Actinic lichen planus is another association, possibly fortuitous. 548 Onset occurs between 20 and 50 years of age. 542 Familial occurrence involving three members of the same family has been reported. 549 Dermoscopy of the pigmented macules on the palms and soles shows a parallel furrow pattern. 550
The changes are similar in all lesions with acanthosis, basal hypermelanosis, and some melanin incontinence with scattered melanophages in the upper dermis. 542 Increased numbers of mildly atypical melanocytes were present in the epidermis and mucosa of one patient. 551 Some large melanosomes have been noted on electron microscopy. 541
The autosomal dominant Peutz–Jeghers syndrome (OMIM 175200) is characterized by the association of gastrointestinal polyposis with the presence of pigmented macules on the buccal mucosa, lips, perioral skin, and sometimes the digits.5.552. and 553. Patients with this syndrome have an increased risk of developing cancer at a relatively young age. There appears to be genetic heterogeneity, although most cases involve the serine/threonine kinase (STK11/LKB1) gene on chromosome 19p13.3. 554 It encodes the protein LKB1 which regulates p53-mediated apoptotic pathways. 555 Mutations in the part of the gene involved in substrate recognition are more frequently associated with malignancies than mutations in the part of the gene involved in ATP binding and catalysis. 556 A case associated with primary melanoma of the rectum has been reported. 557 The Cronkhite–Canada syndrome (OMIM 175500) is also characterized by intestinal polyposis and lentigo-like macules, commonly on the face, extremities, and the palms. 558 The pigmentation tends to be diffuse rather than spotted as in the Peutz–Jeghers syndrome. The etiology is unknown.
Bannayan–Riley–Ruvalcaba (Ruvalcaba–Myhre–Smith) syndrome (OMIM 153480) combines juvenile polyposis coli, macrocephaly, and pigmented macules limited to the shaft and glans of the penis. 561 It results from mutations in the PTEN gene on chromosome 10q23.31. 561 It is allelic with Cowden’s syndrome (see p. 767). 562
Becker’s nevus (melanosis) is usually found in the region of the shoulder girdle of young men as unilateral, hyperpigmented areas of somewhat thickened skin.534. and 563. The front of the chest is another common site, but lesions may occur on any area of the body, 564 including the face. 565 Multiple lesions are rare. 566 Giant bilateral Becker’s nevus is extremely rare. 567 The male to female ratio is anywhere from 4 : 1 to 6 : 1. 568 Hypertrichosis may develop after the pigmentation but is not invariable. 569 A Becker’s nevus is usually acquired in adolescence, but a congenital onset has been recorded,570. and 571. as have familial cases.572. and 573. Its prevalence in a large series of French military recruits was 0.52%. 574 It is more common in young people with fair skin. 575 Occasionally, lesions have been said to follow severe sunburn. Lesional tissue has been found to have an increased level of androgen receptors, suggesting that heightened local androgen sensitivity may result in the hypertrichosis. 576 Various skeletal malformations have been reported in individuals with a Becker’s nevus.577.578. and 579. Other associations have included neurofibromatosis, 580 segmental nevus depigmentosus (a possible example of twin spotting), 581 a connective tissue nevus,582. and 583. acquired superficial angiomatosis, 584 an accessory scrotum, 585 limb deformities, and areolar hypoplasia.586. and 587. Diseases reported to have developed in a Becker’s nevus include eczematous dermatitis and prurigo nodularis, 588 hypohidrosis, 589 acneiform lesions,569.590. and 591. lichen planus, 571 localized scleroderma, 564 and a basal cell carcinoma combined with a melanocytic nevus and a smooth muscle hamartoma. 592
Treatment is usually with laser therapy. Ablative lasers usually leave scarring. Q-switched lasers are used in pigmented variants but several treatments are necessary. The best results (in pigmented lesions) have been obtained with the erbium:YAG lasers.574. and 593.
The epidermal changes are variable but usually there is acanthosis and sometimes mild papillomatous hyperplasia (Fig. 10.6). The changes may resemble those seen in an epidermal nevus (see p. 668), although in Becker’s nevus the elongated rete ridges tend to have flat, rather than pointed tips. There is variable hyperpigmentation of the basal layer with some melanophages in the dermis. Melanocyte proliferation is usually mild and not always obvious in routine sections; special studies have shown a quantitative increase. 594 There is sometimes an increase in the number and size of hair follicles and sebaceous glands. There may be smooth muscle hypertrophy of the arrectores pilorum as well as smooth muscle bundles in the dermis which are not related to cutaneous adnexa.595. and 596. Controversy exists about the relationship of these cases to smooth muscle hamartoma (see p. 860).597. and 598.
Becker’s nevus. (A) The epidermis shows mild papillomatosis and basal hyperpigmentation. (B) The bottom of some of the rete pegs is straight. (H & E)
There is an increase in the number and size of melanosome complexes in the basal and prickle cells of the epidermis with an increase in the number of melanosomes in the complexes. 599 There are also many single collagen fibrils in the dermis.
Dowling–Degos disease (OMIM 179850), also known as reticulate pigmented anomaly of the flexures, is a rare autosomal dominant genodermatosis in which there are spotted and reticulate pigmented macules of the flexures.600. and 601. Recently, a loss-of-function mutation in the keratin 5 (KRT5) gene mapping to chromosome 12q13 (but assigned in one report to 17p13.3602), has been described in this disorder.603. and 604. Whereas mutations affecting the early N-terminal domain of KRT5 appear to be responsible for Dowling–Degos disease, similar mutations in KRT14 produce Naegeli–Franceschetti–Jadassohn syndrome.604. and 605. Less constant features include pigmented pits in the perioral area, localization to the vulva,606. and 607. or axillae, 608 scattered comedo-like lesions,609. and 610. keratoacanthomas, 611 squamous cell carcinomas, 612 hidradenitis suppurativa, 613 and seborrheic keratoses.600.614. and 615. The condition usually develops in early adult life and is slowly progressive. 600 Patients with achromic macules and papules probably constitute a variant of Dowling–Degos disease. 616 Such cases have some overlap features with dyschromatosis symmetrica hereditaria (see p. 292) and dyschromatosis universalis hereditaria.603. and 617.
It is now considered that Haber’s disease,618. and 619. in which there are rosacea-like facies and seborrheic keratosis-like lesions, and reticulate acropigmentation of Kitamura,415.620.621.622.623.624.625. and 626. in which there are reticulate, slightly depressed, pigmented macules on the extensor surface of the hands and feet in association with palmar ‘pits’, are different phenotypic expressions of the same genodermatosis.627.628.629.630.631.632. and 633. Overlap features are sometimes present.634. and 635. A further related entity, characterized by reticulate pigmentation on the face and neck and epidermal cysts on the trunk, has been reported. 636
A further variant is Galli–Galli disease in which patients display prominent acantholytic changes on histology, in addition to clinical and pathological features resembling those of Dowling–Degos disease. 637 Two patients have now been reported with erythematous scaly plaques and lentigo-like macules on the trunk and lower extremities, rather than the reticulate macules of the flexures as usually seen. 638 Mutations in the KRT5 gene, encoding one of the two major basal epidermal keratin intermediate filaments, have now been described in Galli–Galli disease. 605 It remains to be seen whether the acantholysis is related to a specific mutation in this gene.
There are filiform downgrowths of the epidermis and also of the variably dilated pilosebaceous follicles.600. and 639. Small horn cysts and comedo-like lesions are also present. Hyperpigmentation is quite pronounced at the tips of the rete ridges. There is a superficial resemblance to the adenoid form of seborrheic keratosis although the downgrowths are more digitate than in seborrheic keratosis and there is no papillomatosis. In one patient with Dowling–Degos disease, a biopsy from the axilla showed features of Galli–Galli disease with suprabasal lacunae. 603 It has been speculated that acantholysis might be more common in Dowling–Degos disease than previously thought, further confirmation that Galli–Galli disease is the same condition with minor alterations in phenotypic expression. 605 In Kitamura’s disease the appearances resemble those seen in a solar lentigo, with club-shaped elongations of the rete ridges but with intervening epidermal atrophy.628.640. and 641. Dopa-positive melanocytes are increased. 642 Desmocollin 3 was increased in the epidermal rete ridges in one case. 643 Other features include melanin incontinence and a mild to moderate superficial perivascular infiltrate of lymphocytes. In Galli–Galli disease, there are digitate downgrowths of the rete ridges with basal hyperpigmentation. There are suprabasal acantholytic lacunae with a slightly parakeratotic roof but no significant dyskeratosis. 644
Melanosomes are markedly increased in keratinocytes and these may be dispersed through the cytoplasm or loosely aggregated.645. and 646. They are of normal size. Melanocytes are increased in number in Kitamura’s variant. 640 There are many melanosomes in melanocytes, keratinocytes, and melanophages. 642
Hyperpigmentation may follow a number of inflammatory dermatoses, particularly those involving damage to the basal layer. 647 Thus, it may follow various disorders that present a lichenoid reaction pattern, such as lichen planus, lichenoid drug eruptions, and fixed drug eruptions. Prominent hyperpigmentation is almost invariable in the resolving phases of a phytophotodermatitis (see p. 534). Sometimes pigmentation is labeled ‘postinflammatory’ but no etiology is apparent. 648 Specific drugs that may cause postinflammatory and/or increased basal pigmentation include the following: prostaglandin analogues; 649 interferon-α used for hepatitis C infection; 650 the antipsychotic drug olanzapine; 651 hydroxyurea; 652 and calcipotriol. 653
In addition to prominent melanin incontinence there may be normal or increased amounts of melanin in the basal layer of the epidermis. Basal pigmentation is prominent in phytophotodermatitis. The repigmentation observed in lichen sclerosus, following the use of topical tacrolimus involved basal hyperpigmentation and not melanin incontinence. 654 Stem cell factor may produce a similar change at injection sites. 655 If basal pigmentation is markedly reduced, the clinical appearance will be of hypopigmentation. There may also be occasional lymphocytes around vessels in the papillary dermis and a mild increase in fibroblasts and even collagen in the papillary dermis. There is usually no evidence of the underlying dermatosis that resulted in the area of pigmentation.
Prurigo pigmentosa is a rare pruritic dermatosis of unknown etiology in which erythematous papules, characteristically on the back, neck and chest, coalesce to form a reticulate pattern. 656 Vesicular and bullous lesions have been reported. 657 This stage resolves within days, leaving a mottled or reticulate hyperpigmentation.658.659.660. and 661. There is a female predominance. The average age of onset is between 23 and 27 years. 662 Over 300 cases have been reported from Japan, leading to the suggestion that an environmental factor is responsible. 663 It is uncommon in the Western world, although over 50 cases have now been reported.664.665.666. and 667. Ketosis has also been implicated.668.669. and 670. Its association with Helicobacter infection may have been fortuitous. 671 Prurigo pigmentosa may be classed as a postinflammatory melanosis. 672 Minocycline is usually first-line therapy.666.667. and 673. Doxycycline, macrolide antibiotics, isotretinoin, 674 and dapsone have also been used to treat this condition.662.664. and 674.
Böer and Ackerman have reviewed the features of 25 patients and another 178 patients recorded in the literature.656. and 676. They have commented on the rapid course of lesions over a period of one week. In the initial phase there is a superficial perivascular and interstitial infiltrate of neutrophils that are soon scattered through the papillary dermis and epidermis. This is followed by neutrophilic spongiosis and focal epidermal microabscesses, accompanied by ballooning and degeneration (both apoptotic and necrotic) of keratinocytes. This is soon followed by the influx of eosinophils and lymphocytes into the upper dermis. There may be focal lichenoid qualities (Fig. 10.7), while in other cases there is extraordinary vacuolar alteration at the dermoepidermal junction. 676 The epidermis becomes variably hyperplastic, parakeratotic, and hyperpigmented.
Prurigo pigmentosa. There are mild interface changes with some vacuolar change and apoptotic keratinocytes. (H & E)
In the late stages there is prominent melanin incontinence with numerous melanophages in the dermis.
Cutaneous pigmentation which is slate gray or bluish black in color may rarely develop in patients with disseminated malignant melanoma.677. and 678. Although generalized, the pigmentation is often accentuated in areas exposed to the light.
The pathogenesis of the pigmentation is controversial. 678 It has been attributed to epidermal hyperpigmentation, the deposition of melanophages that have circulated in the blood, the presence of scattered melanoma cells within the dermis, 679 and the regression of dermal tumor cells (see p. 747).
The usual finding is the presence of melanin pigment throughout the dermis in perivascular and interstitial melanophages and as free granules. A scant perivascular infiltrate of lymphocytes and sometimes plasma cells may be present. Individual melanoma cells are not usually present.
Dermatopathia pigmentosa reticularis (OMIM 125595) combines generalized reticulate pigmentation with nail dystrophy and partial alopecia.680.681.682. and 683. Macules of hypopigmentation may develop at a later stage. There are similarities to the Naegeli–Franceschetti–Jadassohn syndrome, with which it is allelic, although the associated features are different. 684 Both conditions are autosomal dominant due to mutations in the keratin-14 (KRT14) gene on chromosome 17q12–q21. 684
Naegeli–Franceschetti–Jadassohn syndrome (Naegeli syndrome – OMIM 161000) is an extremely rare, autosomal dominant ectodermal dysplasia combining dark brown, reticulate pigmentation of the trunk and limbs with diffuse or punctate hyperkeratosis of the palms and soles.685. and 686. Hypohidrosis, enamel hypoplasia, and nail dystrophy may also be present. Incomplete forms or variations have been reported;687. and 688. the term ‘hereditary diffuse hyperpigmentation’ was used for one such case. 689 The gene for this syndrome, the keratin-14 (KRT14) gene, maps to chromosome 17q11.2–q21 (also given as 17q12–q21).684. and 690. As such it is allelic to dermatopathia pigmentosa reticularis. 684
This genodermatosis is one of the many that may present with reticulate, patchy, and mottled pigmentation of the neck. A review of all such dermatoses, congenital and acquired, was published some years ago.691. and 692.
Incontinentia pigmenti (OMIM 308300) is an uncommon, multisystem genodermatosis with cutaneous, skeletal, ocular, neurological, dental, and other abnormalities.693.694.695. and 696. The ocular and neurological changes are sometimes the dominant clinical features of the disorder.697.698.699.700. and 701. The cutaneous lesions evolve through vesiculobullous, verrucous, and pigmentary stages, but in a small number of individuals pigmentation is the first manifestation. This occurs when there is reduced expression of the phenotype. 702 The vesiculobullous lesions, accompanied by erythematous areas, are present at birth or soon after, in a linear arrangement on the extremities and lateral aspects of the trunk. Skin lesions tend to be patterned along Blaschko’s lines. 700 Rarely, they are papular in type. 703 Vesicular recurrences, later in life, are rare.704.705.706. and 707. The verrucous lesions evolve some weeks or months later and resolve spontaneously to give atrophy, depigmentation or both. In the third stage, which has a peak onset around 3–6 months, there are streaks and whorls of brown to slate gray pigmentation, often asymmetrically distributed on the trunk and sometimes on the extremities.694. and 708. The pigmentation, which is not necessarily in areas of the earlier lesions, progressively fades at about puberty. Areas of hyperpigmentation, sometimes associated with verrucous plaques, may remain. 709 Uncommonly, streaks of hypopigmentation are the predominant feature;710.711.712. and 713. they are usually found in adulthood but may develop earlier.714. and 715.
Other cutaneous manifestations include alopecia, rarely of a whorled scarring type, 716 woolly hair nevus, 717 nail dystrophy,718. and 719. and painful subungual tumors which may involve several fingers and sometimes toes.720. and 721. These keratotic tumors have an onset in late adolescence and may involute spontaneously.722. and 723. Several cases of incontinentia pigmenti have been associated with cancer in childhood. 724 One case was associated with neonatal herpes simplex infection. 725 Peripheral eosinophilia is often present. 726
Incontinentia pigmenti is a chromosomal instability disorder which is inherited as an X-linked dominant gene that usually causes the death in utero of affected males.712.717.724. and 727. Heterozygous females survive owing to functional mosaicism. 728 Two gene loci have been identified for this disease: Xp11.21 and Xq28, 704 although recent studies and OMIM list only Xq28. 729 Mutations in the IKK-gamma gene (IKBKG), also called NEMO, which maps to Xq28, cause incontinentia pigmenti. The NEMO/IKKγ gene controls the nuclear factor κB (NF-κB) signaling pathway which controls various cytokines and chemokines, and protects cells against apoptosis.730. and 731. In the dermatological literature, NEMO (nuclear factor-κB essential modulator) is the preferred designation for this gene.728.732. and 733. Loss of function mutation of NEMO accounts for 80% of cases. Hypomorphic mutations of NEMO result in incontinentia pigmenti in heterozygous females and in hypohidrotic ectodermal dysplasia associated with severe immunodeficiency (OMIM 300291) which is allelic. 728 The small number of males (now close to 50) reported with the condition726.729. and 734. may represent genetic heterogeneity mutations although several cases have shown mosaicism for a deletion of the NEMO gene.694.728. and 729. In one boy, XXY mosaicism was present. 735 Father-to-daughter transmission has been recorded. 736
Several patients have had defects in neutrophil chemotaxis and lymphocyte function.737.738.739. and 740. Leukotriene B4 has been demonstrated in extracts of the crusted scales from vesiculobullous lesions and this may have an important role in the chemotaxis of eosinophils into the epidermis. 741 More important is the activation of eotaxin, a direct consequence of the genetic mutation, which is a potent eosinophil-selective chemokine. It is increased in the skin during the vesiculobullous stage. 697
It has been postulated that the manifestations of incontinentia pigmenti can be explained as an autoimmune attack on ectodermal clones expressing an abnormal surface antigen742 or as premature (programmed) cell death in defective ectodermal clones. 743 Apoptosis is increased as a direct consequence of the mutation.
The first stage of incontinentia pigmenti is characterized by eosinophilic spongiosis; that is, spongiosis progressing to intraepidermal vesicle formation with prominent exocytosis of eosinophils into and around them (Fig. 10.8). A few basophils are also present. 744 The erythematous areas show only minimal spongiosis, but there is still prominent exocytosis of eosinophils. There are occasional dyskeratotic cells with eosinophilic hyaline cytoplasm in the epidermis adjacent to the vesicles. 717 The superficial dermis contains an infiltrate of eosinophils and some mononuclear cells. Eosinophil granule major basic protein is also present. 745
Incontinentia pigmenti (first stage). Numerous eosinophils extend into the epidermis. Spongiosis is mild in this field. (H & E)
In the verrucous stage, there are hyperkeratosis, acanthosis, mild irregular papillomatosis, and numerous dyskeratotic cells (Fig. 10.9). Some macrophages migrate into the epidermis; on electron microscopy, these have been shown to phagocytose the dyskeratotic cells as well as melanosomes. Inflammatory cells are quite sparse. In the third stage there is pronounced melanin incontinence. Pale scarred areas may be found on the lower part of the legs; these show a reduction in the number of melanocytes and some increase in dermal collagen. 746
Incontinentia pigmenti (verrucous stage). There are many dyskeratotic cells within the epidermis. (H & E)
The subungual lesions show hyperkeratosis, verrucous or pseudoepitheliomatous hyperplasia, and dyskeratotic cells at all levels of the epidermis.720.722. and 723. Neighboring keratinocytes may form whorls around the dyskeratotic cells.
This X-linked disorder (OMIM 301220) is characterized by cutaneous pigmentation mimicking incontinentia pigmenti, associated with failure to thrive, chronic pulmonary disease, hypohidrosis, and coarse hair. 750 The gene maps to Xp21 near the dystrophin locus. 750 Although amyloid was found in the papillary dermis of the original case, it has not been found in subsequent cases. At one stage, this condition was called familial cutaneous amyloidosis. 750
Affected skin shows mild hyperkeratosis, basal hyperpigmentation, and numerous dermal melanophages. Occasional apoptotic keratinocytes are also present. 750 No amyloid has been present, other than in the original report.
Localized hyperpigmentation may develop at sites of chronic friction.751. and 752. This condition must be distinguished from macular amyloidosis, which clinically it resembles. Frictional melanosis usually occurs over bony prominences, following prolonged and repetitive friction. In some countries the use of a scrub pad (loofah) has been implicated. 753 It is more common in patients who have skin phototype III–V. 754
Sock-line hyperpigmentation is a related condition due to trauma from the elastic bands often present in the upper portion of socks.755. and 756. It has also been likened to amniotic bands of infancy.
Notalgia paresthetica is a sensory neuropathy involving the posterior primary rami of thoracic nerves T2 to T6 and presenting as a localized area of pruritus of the back.757.758. and 759. The affected region is sometimes lightly pigmented and composed of groups of small tan macules. Similar cases have been reported in the literature as ‘peculiar spotty pigmentation’760 and ‘idiopathic pigmentation of the upper back’. 761 Clinically, notalgia paresthetica resembles macular amyloidosis, a condition which in one report required ultrastructural examination to confirm the presence of amyloid, as histochemical tests were negative. 762
The symptoms may result from an increase in sensory epidermal innervation in affected skin. 763 More likely is the role played by degenerative changes in the spine, leading to spinal nerve impingement.764. and 765. In one series of 43 patients, 60.7% had spinal changes deemed to be relevant. 766
Treatment with botulinum toxin type A, by intradermal injections, was successful in one small series of two patients. 767
There is melanin pigment in macrophages in the upper dermis, sometimes accompanied by mild hyperpigmentation of the basal layer (Fig. 10.10).761. and 768. The pattern is that of mild postinflammatory pigmentation. In several reported cases, scattered degenerate keratinocytes were present within the epidermis.757. and 763. Although no amyloid was seen in two combined series of 24 cases, it is often present.765. and 769. It is possible that amyloid forms in chronic lesions as a consequence of prolonged scratching.
Notalgia paresthetica. There are numerous melanophages, particularly around vessels in the superficial plexus. No amyloid was present. (H & E)
Immunohistochemistry with neural and neuropeptide markers did not reveal a significant difference between lesional skin and non-lesional skin. 769
Although ‘ripple’ pigmentation is usually regarded as a feature of macular amyloidosis, it has also been described on the neck in almost 2% of individuals with atopic dermatitis of long standing.770. and 771.
‘Terra firma-forme’ dermatosis is a relatively common condition that usually affects the neck of children. It has also been reported as dermatitis neglecta (see p. 526). It has the appearance of a dirty brown mark that cannot be washed off with soap but is easily removed with alcohol.772.773. and 774. It can often be mistaken for acanthosis nigricans and other conditions. It appears to be caused by disordered keratinization.
There is mild acanthosis and orthokeratosis with numerous keratin globules in the stratum corneum.