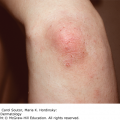
4 The complex but orderly processes of keratinization, and of cell cohesion and proliferation within the epidermis, have been described in Chapter 2. As they proceed, the living keratinocytes of the deeper epidermis change into the dead corneocytes of the horny layer, where they are stuck together by intercellular lipids. They are then shed in such a way that the surface of the normal skin does not seem scaly to the naked eye. Shedding balances production, so that the thickness of the horny layer does not alter. However, if keratinization or cell cohesion is abnormal, the horny layer may become thick or the skin surface may become dry and scaly; this impairs barrier function, which if severe can lead to excessive water loss, dehydration and, in extreme cases, death. Such changes can be localized or generalized. In this chapter we describe a variety of skin disorders that have as their basis a disorder of keratinization. The term Mendelian disorders of cornification (MEDOC) has recently been used to classify these conditions. During the last few years the molecular mechanisms underlying many of these have become clearer, including abnormal genetic coding for keratins, the enzymes involved in cell cohesion in the horny layer and the molecules that are critical in the signalling pathway governing cell cohesion in the spinous layer. The word ichthyosis comes from the Greek word for a fish. It is applied to disorders that share, as their main feature, a dry rough skin with marked scaling but no inflammation. Strictly speaking, the scales lack the regular overlapping pattern of fish scales, but the term is usefully descriptive and too well entrenched to be discarded. Much information on icthyoses is coming to light from national patient registers, both in the United Kingdom and the United States. In 2009, experts in the field of ichthyosis met to agree on an international consensus classification (see Further reading). This supersedes older nomenclature and as such has been adopted in this chapter. The genetics underlying the ichthyoses is complex; to date there are 36 forms of inherited ichthyosis, including conditions primarily affecting the skin, and rarer ‘syndromic’ associations involving other organs. Over 25 genes have now been implicated, with multiple mutations on each gene. Recent research has focused on the molecular pathways by which the genetic defects produce the ichthyosiform phenotype. This chapter reviews the more commonly encountered ichthyoses and provides suggestions for further reading on the rarer forms. Inherited as an autosomal semidominant disorder, this condition is common, affecting about 1 in 250 people in the United Kingdom. Mutations in the filaggrin gene lead to a loss or reduction of profilaggrin, the major component of the keratohyalin granule. Profilaggrin is cleaved to filaggrin, which in turn is responsible for aggregating keratin filaments in the cornified cell envelope. The breakdown of filaggrin results in the formation of filaggrin degradation products, which reduce transepidermal water loss. The reduction in keratohyalin granules gives icthyosis vulgaris its characteristic histology; a paucity or absence of the granular layer of the epidermis. Mutant alleles of the filaggrin gene have a frequency of 4% in European populations, which accounts for it being such a common disorder. Heterozygotes have milder disease than compound heterozygotes and homozygotes. The dryness is usually mild and symptoms are few. The scales are small and branny, being most obvious on the limbs and least obvious in the major flexures. The skin creases of the palm may be accentuated. Keratosis pilaris (p. 48) is often present on the limbs. The skin changes are not usually present at birth but develop over the first few years of life. Some patients improve in adult life, particularly during warm weather, but the condition seldom clears completely. The already dry skin chaps in winter and is easily irritated by degreasing agents. This should be taken into account in the choice of a career. Ichthyosis of this type is apt to appear in a stubborn combination with atopic eczema, as mutations in the filaggrin gene are strong predisposing factors for atopic eczema (p. 88). It can usually be distinguished from less common types of ichthyosis on the basis of the pattern of inheritance and the type and distribution of the scaling. Mild variants may be difficult to differentiate from the xerosis of atopic eczema, which is not surprising given their common genetic basis. None are usually needed. This is palliative. The dryness can be helped by the regular use of emollients, which are best applied after a shower or bath. Emulsifying ointment, soft white paraffin, E45 and Unguentum Merck are all quite suitable (Formulary 1, p. 397) and the selection depends on the patient’s preference. Many find proprietary bath oils and creams containing humectants such as glycerin, urea or lactic acid helpful (Formulary 1, p. 397). This less common type of ichthyosis is inherited as an X-linked recessive trait and therefore, in its complete form, is seen only in males, although some female carriers show mild scaling. The condition affects about 1 in 6000 males in the United Kingdom and is associated with a deficiency of the enzyme steroid sulfatase, which hydrolyses cholesterol sulfate. The responsible gene has been localized to the end of the short arm of the X chromosome, at Xp 22.3 (see Chapter 24). In contrast to the delayed onset of the dominantly inherited ichthyosis vulgaris, scaling appears early, often soon after birth, and always by the first birthday. The scales are larger and browner (Figure 4.1), involve the neck, and to a lesser extent the popliteal and antecubital areas, as well as the skin generally. The palms and soles are normal. There is no association with atopy or keratosis pilaris. The condition persists throughout life. Figure 4.1 Ichthyosis: large rather dark scales suggest recessive X-linked ichthyosis. Affected babies may be born after a prolonged labour. Corneal opacities may appear in adult life. Kallmann’s syndrome (hypogonadotrophic hypogonadism and anosmia) is caused by the deletion of a part of the X chromosome that includes the gene for X-linked recessive ichthyosis, which is therefore one of its features. Neurological defects may also occur in this contiguous gene disorder. This is as for ichthyosis vulgaris. It is helpful to remember that only males are affected. Bear Kallmann’s syndrome in mind if there are other congenital abnormalities. None are usually needed. Steroid sulfatase gene deletions can be identified by fluorescence in situ hybridization (FISH; see Chapter 24). Electron microscopy shows retained corneodesmosomes within the stratum corneum (see Chapter 2). Oral aromatic retinoids are probably best avoided. Topical measures are as for ichthyosis vulgaris. There are three major types of autosomal recessive congenital ichthyosis: lamellar ichthyosis, congenital ichthyosiform erythroderma and harlequin ichthyosis. The term ‘collodion baby’, often associated with these conditions, is a description and not a diagnosis. The bizarre skin changes are seen at birth. At first the stratum corneum is smooth and shiny, and the skin looks as though it has been covered with cellophane or collodion. Its tightness may cause ectropion and feeding difficulties. The shiny outer surface is shed within a few days leaving behind red scaly skin. This is most often a result of congenital ichthyosiform erythroderma, less commonly lamellar ichthyosis, and is very severe in harlequin ichthyosis. Collodion babies may have problems with temperature regulation and high water loss through the skin in the early days of life, best dealt with by the use of a high humidity incubator. Regular application of a greasy emollient also limits fluid loss and makes the skin supple.
Disorders of Keratinization
The ichthyoses
Common ichthyoses
Ichthyosis vulgaris
Cause
Presentation
Clinical course
Complications
Differential diagnosis
Investigations
Treatment
Recessive X-linked ichthyosis
Cause
Presentation and course
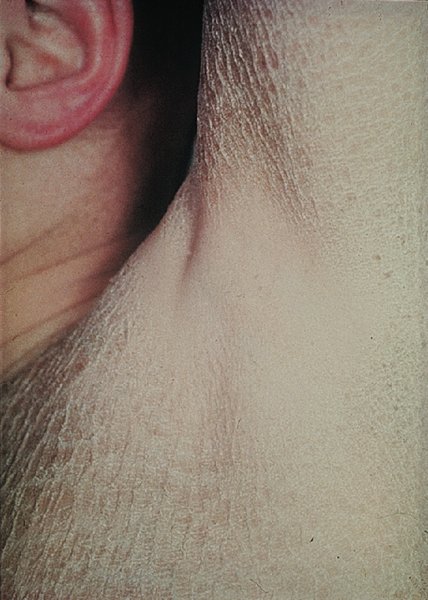
Complications
Differential diagnosis
Investigations
Treatment
Autosomal recessive congenital ichthyoses
Lamellar ichthyosis and congenital ichthyosiform erythroderma
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


