Introduction304
INTRODUCTION
Normal collagen
Composition of collagen1
Types of collagen3.4.5. and 6.
Metabolism of collagen
Categorization of collagen disorders
SCLERODERMA
LOCALIZED SCLERODERMA
Morphea
Linear scleroderma
Histopathology22
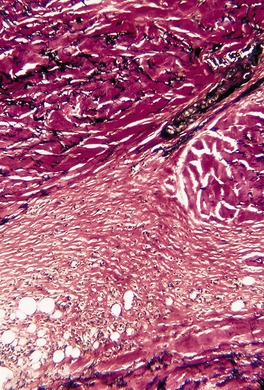
Fig. 11.1
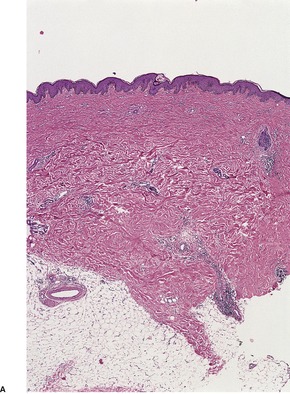
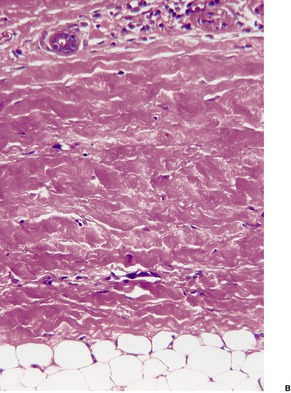
Fig. 11.2
SYSTEMIC SCLERODERMA
Diffuse systemic scleroderma
Limited systemic scleroderma
Pathogenesis of scleroderma
Treatment of scleroderma
Histopathology197.297.298. and 299.
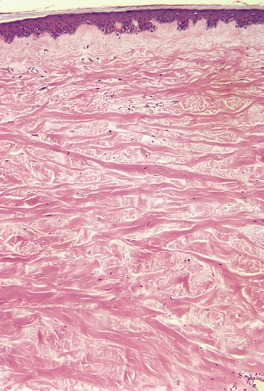
Fig. 11.3
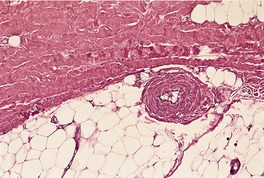
Fig. 11.4
MIXED CONNECTIVE TISSUE DISEASE
Histopathology
EOSINOPHILIC FASCIITIS
Histopathology330. and 364.
ATROPHODERMA
Histopathology
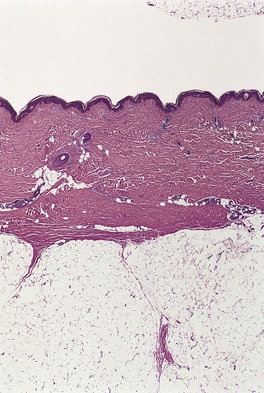
Fig. 11.5
Electron microscopy
SCLERODERMOID DISORDERS
SCLERODERMOID GRAFT-VERSUS-HOST DISEASE
Histopathology
STIFF-SKIN SYNDROME
Histopathology
WINCHESTER SYNDROME
Histopathology
GEMSS SYNDROME
Histopathology
PACHYDERMOPERIOSTOSIS
Histopathology436. and 453.
PACHYDERMODACTYLY
ACRO-OSTEOLYSIS
Histopathology461
CHEMICAL- AND DRUG-RELATED DISORDERS
Histopathology
PARANEOPLASTIC PSEUDOSCLERODERMA
Histopathology
NEPHROGENIC SYSTEMIC FIBROSIS
LICHEN SCLEROSUS ET ATROPHICUS
Treatment of lichen sclerosus et atrophicus
Histopathology634
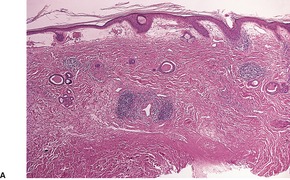
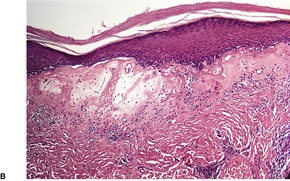
Fig. 11.6
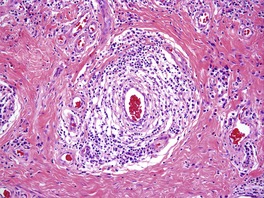
Fig. 11.7
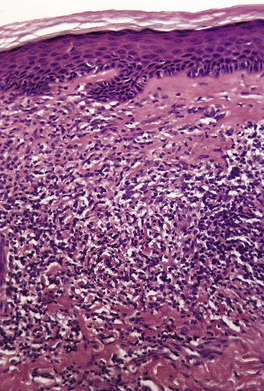
Fig. 11.8
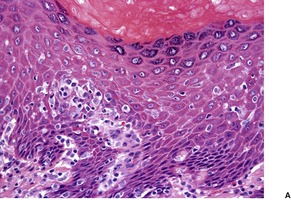
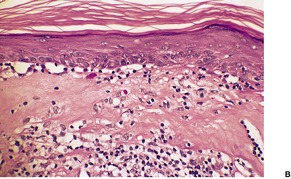
Fig. 11.9
Electron microscopy
POST-STRIPPING CUTANEOUS SCLEROSIS
Histopathology
OTHER HYPERTROPHIC COLLAGENOSES
CONNECTIVE TISSUE NEVI
Collagenoma
Histopathology
Shagreen patch
Histopathology719
WHITE FIBROUS PAPULOSIS OF THE NECK
Histopathology
HYPERTROPHIC SCARS AND KELOIDS
Histopathology739. and 798.
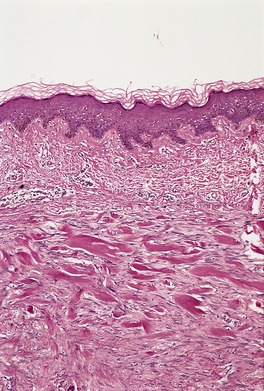
Fig. 11.10
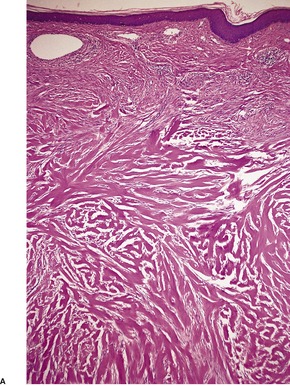
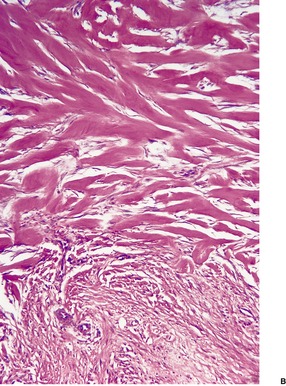
Fig. 11.11
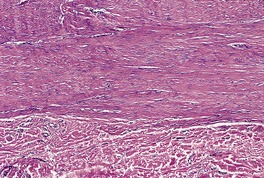
Fig. 11.12
Electron microscopy
STRIAE DISTENSAE
Histopathology830.831. and 832.
FIBROBLASTIC RHEUMATISM
Histopathology
COLLAGENOSIS NUCHAE
Histopathology
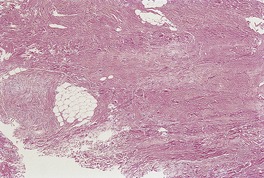
Fig. 11.13
LIPODERMATOSCLEROSIS
WEATHERING NODULES OF THE EAR
Histopathology856
ATROPHIC COLLAGENOSES
APLASIA CUTIS CONGENITA
Type
Clinical features
I
Scalp ACC without multiple abnormalities
II
Scalp ACC with limb reduction abnormalities
III
Scalp ACC with epidermal and organoid nevi associated with corneal opacities and psychomotor retardation
IV
ACC overlying embryological abnormalities, neural or omphalocele
V
ACC with fetus papyraceus or placental infarct
VI
ACC associated with epidermolysis bullosa
VII
ACC localized to extremities without associated abnormalities
VIII
ACC due to teratogens, virus, or drug
IX
ACC associated with malformation syndromes such as trisomy 13, ectodermal dysplasia, or Goltz syndrome
Histopathology877
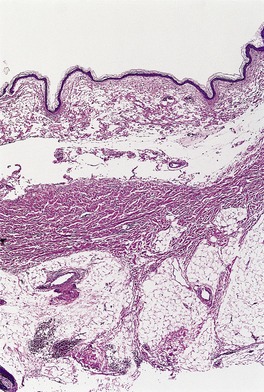
Fig. 11.14
FOCAL DERMAL HYPOPLASIA
Histopathology917.930. and 944.
Electron microscopy
FOCAL FACIAL DERMAL DYSPLASIA
Histopathology
PSEUDOAINHUM CONSTRICTING BANDS
Histopathology962
KERATOSIS PILARIS ATROPHICANS
Histopathology
CORTICOSTEROID ATROPHY
Histopathology977. and 983.
Electron microscopy
LINEAR ATROPHODERMA OF MOULIN
ACRODERMATITIS CHRONICA ATROPHICANS
RESTRICTIVE DERMOPATHY
Histopathology
PERFORATING COLLAGENOSES
REACTIVE PERFORATING COLLAGENOSIS
Histopathology1008.1012. and 1017.
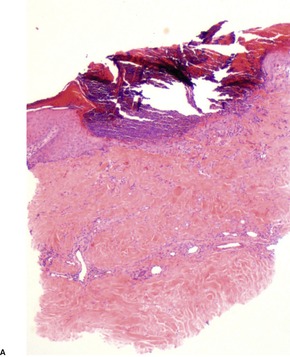
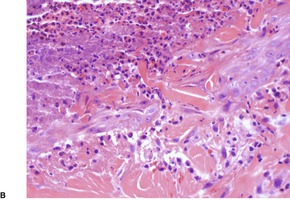
Fig. 11.15
PERFORATING VERRUCIFORM ‘COLLAGENOMA’
Histopathology1070
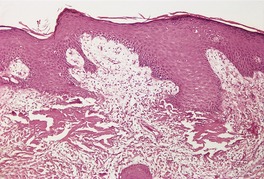
Fig. 11.16
CHONDRODERMATITIS NODULARIS HELICIS
Histopathology1090. and 1091.
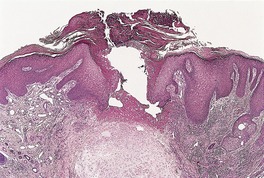
Fig. 11.17
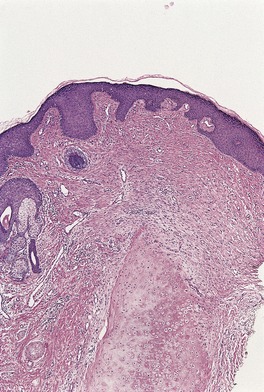
Fig. 11.18
VARIABLE COLLAGEN CHANGES
EHLERS–DANLOS SYNDROME
New name
Former category (traditional classification)
Classic type
Hypermobility type
Benign hypermobile (type III)
Vascular type
Arterial-ecchymotic (type IV)
Kyphoscoliosis type
Ocular-scoliotic (type VI)
Arthrochalasis type
Arthrochalasis congenita (types VIIA and B)
Dermatosparaxis type
Human dermatosparaxis (type VIIC)
Other forms
Histopathology
Subtypes of Ehlers–Danlos syndrome (traditional classification)
Type I (gravis)
Type II (mitis)
Type III (benign hypermobile)
Type IV (arterial, ecchymotic)
Type V (X-linked)
Type VI (ocular/kyphoscoliosis)
Type VII (arthrochalasis congenita)
Type VIII (periodontal)
Type IX
Type X (fibronectin)
Type XI (familial joint instability syndrome)
Other types
OSTEOGENESIS IMPERFECTA
Histopathology
MARFAN’S SYNDROME
Histopathology16
SYNDROMES OF PREMATURE AGING
WERNER’S SYNDROME (ADULT PROGERIA)
Histopathology
PROGERIA
Histopathology1193. and 1199.
ACROGERIA
Histopathology1165.1201. and 1202.
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Disorders of collagen
Sclerodermoid disorders311
Sclerodermoid graft-versus-host disease312
Stiff-skin syndrome312
Winchester syndrome312
GEMSS syndrome312
Pachydermoperiostosis312
Pachydermodactyly313
Acro-osteolysis313
Chemical- and drug-related disorders313
Paraneoplastic pseudoscleroderma313
Nephrogenic systemic fibrosis313
Lichen sclerosus et atrophicus313
Post-stripping cutaneous sclerosis316
Atrophic collagenoses321
Collagen is the major structural constituent of mammalian connective tissues. 1 It accounts for well over 70% of the dry weight of the skin. 2 There are at least 10 genetically distinct types of collagen and it is the relative content of these different collagen types, as well as the amount of elastic tissue and non-structural constituents such as the proteoglycans, that determines the specific biomechanical properties of the various connective tissues.3. and 4.
Before discussing the disorders of collagen in the skin, a brief account will be given of the composition, types, and metabolism of collagen.
The structural collagens – types I, II, and III – are composed of three polypeptide chains, called alpha chains, each of which contains approximately 1000 amino acid residues, one-third of which are glycine. Proline and hydroxyproline are other important amino acids, constituting up to 20% of the amino acids. The sequence and composition of amino acids differ in the alpha chains of the various collagens. 4 Each of the alpha chains is coiled in a helix, and the three chains which together constitute a collagen molecule are in turn coiled on each other to form a triple helical structure. Short non-helical extensions are found at both ends of the molecule at the time it is secreted into the tissues. These extensions are soon cleaved from the procollagen molecules by two different proteases. This produces a shorter molecule which, by lateral and longitudinal association with others, produces collagen fibrils. At the same time, oxidation of lysyl and hydroxylysine residues results in the formation of stable crosslinks that give tensile strength to the collagen.
As mentioned above, at least 10 genetically distinct collagens have now been characterized, and at least 20 distinct genes encode the subunits of the various types of collagen. 7 Two of the three structural collagens, types I and III, are important constituents of the skin, while type IV collagen is an important constituent of the basement membrane (see p. 141). Only small amounts of the other collagen types are found in the skin. 3
Type I collagen is the most abundant collagen in the dermis. 3 It comprises about 80% of dermal collagen and plays a major role in providing tensile strength to skin. 8 It is composed of two identical alpha chains and a third chain of different amino acid composition. The genes for these different chains are thought to be on chromosomes 17 and 7 respectively.
Type III collagen constitutes approximately 50% of fetal skin but less than 20% of adult skin. 2 It is also present in internal organs. This collagen type is composed of three identical alpha chains. Type III collagen is believed to accommodate the expansion and contraction of tissues such as blood vessels and viscera. 9 Reticulin fibers may represent type III collagen. The synthesis of type III collagen is controlled by the COL3A1 gene on chromosome 2q31–q32.
Type IV collagen has a honeycomb or reticular pattern in contrast to the fibrillar pattern of the other major collagen types.10. and 11. It is an important constituent of the lamina densa of the basement membrane.10. and 11. Type V collagen is a low-abundance fibrillar collagen that is coexpressed with collagen I in many tissues and forms with it heterotypic fibrils. 12 Collagen V appears to play a crucial role in the assembly of these heterotypic fibers and in regulating their diameter. 12 An abnormality has been found in some patients with Ehlers–Danlos syndrome type I. Type VII collagen is found in the sublamina densa region of the basement membrane zone forming the anchoring filaments (see p. 141). Type XVI collagen, a member of the fibril-associated collagens with interrupted triple helices, localizes preferentially in the papillary dermis. It is also found in some sclerotic processes, such as scleroderma. 13 Type XVII collagen is the bullous pemphigoid antigen.
The metabolism of collagen is a complex process involving a balance between its synthesis and its degradation. 14 There are numerous steps involved in the synthesis of collagen, and the regulatory mechanisms are not fully understood. Procollagen is formed in the rough endoplasmic reticulum of fibroblasts. After passing through the Golgi apparatus it is transported to the cell surface and secreted into the interstitium of the connective tissue. 14 Here occur cleavage of the terminal extensions of procollagen and the subsequent crosslinking of molecules to form stable collagen. 15 Collagen appears to be turned over continuously; its degradation is brought about by collagenase. It is remarkably resistant to proteolysis by most tissue proteinases. 14
Various substances can interfere with the synthesis of collagen: the most important are the corticosteroids, which appear to act at several levels in the biosynthetic pathway. 15
Although the various disorders of connective tissue have been assigned to a particular chapter of this volume on the basis of which element is most affected, it must be emphasized that an alteration in one component of connective tissue may influence the synthesis, deposition, and structure of other components. 16 For instance, alterations in the elastic tissue and proteoglycan composition of the dermis may be found in some of the primary disorders of collagen.
The following categories will be considered, although it is acknowledged that the allocation of some of the disorders to a particular section is somewhat arbitrary:
• scleroderma
• sclerodermoid disorders
• other hypertrophic collagenoses
• atrophic collagenoses
• perforating collagenoses
• variable collagen changes
• syndromes of premature aging.
It should be noted that diseases such as systemic lupus erythematosus and polyarteritis nodosa, which have been regarded in the past as ‘collagen diseases’, are not included in this chapter as they are not disorders of collagen in the strict sense. They are discussed with their appropriate tissue reaction pattern.
The term ‘scleroderma’ refers to a group of diseases in which there is deposition of collagen in the skin and sometimes other organs as well. It may occur as a localized cutaneous disease in which the disorder of connective tissue is limited to the skin and sometimes structures beneath the affected skin, or it may occur as a systemic disease in which cutaneous lesions are accompanied by Raynaud’s phenomenon and variable involvement of other organs.17.18.19. and 20.
This classification of scleroderma will be used in the account that follows:
1. localized scleroderma
morphea and variants
linear scleroderma
2. systemic scleroderma
diffuse form (progressive systemic sclerosis)
3. mixed connective tissue disease
4. eosinophilic fasciitis
5. atrophoderma (of Pasini and Pierini).
Localized scleroderma is the most common form of scleroderma. 21 It generally occurs in children and young adults and there is a female preponderance.22. and 23. Neonatal onset has been recorded. 24 There is no visceral involvement or Raynaud’s phenomenon and it usually has a self-limiting course. Progression to or coexistence of the systemic form is rare.22. and 25. Antinuclear antibodies are uncommon except in the linear form. 26
Morphea is the commonest form of scleroderma. Usually it presents on the trunk or extremities as one or several indurated plaques with an ivory center and a violaceous border (the ‘lilac ring’). Lesions confined to the breast have been reported. 27 Irregular areas of hyperpigmentation or hypopigmentation may be present within the lesion. 28 Other clinical forms include guttate, generalized, subcutaneous, keloidal, and bullous types.22.29.30. and 31. Occasionally, more than one type is present in the same individual.
Guttate morphea consists of small, pale, slightly indurated lesions on the upper part of the trunk which may resemble lichen sclerosus et atrophicus. 32 In the rare generalized morphea there are large plaque-like lesions, often with vague symmetry, involving the trunk and extremities. 33 Atrophy and fibrosis of the deep tissues may lead to crippling deformities. Ulceration and calcification may also develop in some of the lesions. 32Disabling pansclerotic morphea is an aggressive variant of the generalized type and is of early onset.34. and 35. Lesions may develop in adult life.36. and 37. Lesions may extend circumferentially on an extremity leading to massive pansclerosis and atrophy.32. and 38. Peripheral eosinophilia and mild non-progressive visceral changes are sometimes present in this variant. 35 Cutaneous squamous cell carcinoma is a rare complication of long-standing lesions.39. and 40.Subcutaneous (deep) morphea (morphea profunda) consists of one or more ill-defined, deep sclerotic plaques on the abdomen, sacral area, or extremities; its progression is slow and usually relentless.41.42.43.44. and 45. Solitary lesions may not progress. 46 Rarely, the lesions have a linear arrangement. 47 Deep morphea has developed at the site of a previous vaccination. 48Keloidal (nodular) morphea may be part of this spectrum, although the nodules may also be in the dermis, clinically resembling a keloid.22.49.50.51. and 52. Nodules are a rare finding in systemic and linear scleroderma.53.54.55.56.57. and 58.Bullous lesions may rarely complicate both localized and systemic scleroderma.29. and 59. They may present initially as acquired unilateral edema. 60 In some cases, bullous morphea represents the secondary appearance of bullous lichen sclerosus et atrophicus on a lesion of morphea.61. and 62.Superficial morphea is a recently described variant that differs in its clinical and histological presentation from classic morphea. 63 There is minimal to no induration of patches that are hypo- or hyperpigmented. 63 Males are uncommonly affected. 64 There are no associated symptoms. It has been suggested that superficial morphea is atrophoderma of Pasini–Pierini.65. and 66.
Lesions of morphea may coexist with lichen sclerosus et atrophicus (see p. 316) 67 and there are isolated reports of localized sclerodermatous lesions occurring in association with elastosis perforans serpiginosa (‘perforating morphea’), 68 granuloma annulare, 69 Hashimoto’s thyroiditis, 70 multiple myeloma, 71 a herpes zoster scar, 72 xanthomatosis, 73 a congenital hypopigmented plaque of unknown type, 74 discoid lupus erythematosus, 75 subacute lupus erythematosus, 76 and the presence of the so-called ‘lupus anticoagulant’ (see p. 201). 77 Generalized morphea has been reported in a patient with Felty’s syndrome. 78 Bronchiolitis obliterans developed in another patient. 79
The etiology of morphea is controversial. Antibodies and lymphoproliferative responses80 to Borrelia burgdorferi, the cause of Lyme disease, have been detected in a significant number of patients with morphea in Austria and some other parts of Europe.81.82. and 83. Spirochetes have been demonstrated in tissue sections in some cases using a modified silver stain, 84 an avidin-biotin immunoperoxidase system85. and 86. or techniques using the polymerase chain reaction (PCR).87. and 88. Organisms have also been detected by focus-floating microscopy. 89 However, these findings have not been confirmed in most other countries90.91. and 92. and this leads to the view that in most instances there is no association between B. burgdorferi infection and morphea.93.94.95.96.97.98. and 99. Another explanation is that certain genotypes of Borrelia found only in parts of Europe and Japan are responsible, as is the situation with lichen sclerosus et atrophicus. 100 However, this explanation is not confirmed by a recent study from Germany which found no evidence of Borrelia by PCR in lesional skin of 33 patients with morphea. 101 It has also been suggested that a new spirochetal agent unrelated to B. burgdorferi may be the causative agent in some countries. 102 The increased collagen synthesis by fibroblasts in morphea may result from lymphokines released by the inflammatory cell infiltrate. 103 Transforming growth factor β (TGF-β) is increased in lesional skin. 104 Serum levels of procollagen type I carboxyterminal propeptide are increased in patients with localized and systemic scleroderma. 105 Insulin-like growth factor (IGF)-I, which is a profibrotic compound, is increased in lesional and non-lesional skin of patients with morphea. 106 Features of autoimmunity are also present in localized scleroderma, with a high percentage of patients having antinucleosome and antihistone antibodies.107.108.109. and 110. Anti-agalactosyl antibodies are present in about 20% of patients, and they can be an indicator of disease severity. 111 Antinuclear antibodies are uncommon except in the linear form; 26 antibodies to single-stranded DNA are sometimes present, particularly in generalized morphea.112. and 113. Rheumatoid factor is present in about 20% of cases.114. and 115. Increased levels of circulating ICAM-1 are present; the levels correlate with the number of lesions and the area involved. 116 Whereas TGF-β is known as a profibrotic cytokine which may have a role in the pathogenesis of scleroderma, decorin reduces TGF-β levels and has antifibrotic properties. Decorin levels are increased in the early stages of scleroderma, particularly the systemic form. 117
Morphea has developed, in a few instances, at the site of previous radiotherapy (radiation port scleroderma).118.119.120.121. and 122. There is a marked up-regulation of collagen synthesis following radiotherapy. 123 Morphea has also been reported in a patient taking the semisynthetic ergot alkaloid bromocriptine, a drug that has been associated with pulmonary fibrosis. 124 It has followed the use of balicatib, a cathepsin K inhibitor. 125 It has also developed adjacent to the site of a leaking silicone-gel breast implant, 126 as a tattoo reaction, 127 and in areas of chronic venous insufficiency. 128
Therapy using various PUVA-related protocols has been used with some success.129. and 130. It has been used with oral retinoids. 131 UVA1 phototherapy is also an effective therapy.132. and 133. Low-dose UVA1 phototherapy can also be given with calcipotriol ointment. 134 Calcipotriol can also be used with corticosteroids. 135 The effects of UVA are mainly restricted to the epidermis and dermis, restricting its use in deep forms of the disease. Photodynamic therapy has also been trialed for the treatment of morphea. 136 Corticosteroids remain the initial treatment of choice for localized lesions. 137 Pulsed high-dose corticosteroids, with low-dose methotrexate, have been given to treat severe cases of localized scleroderma.138. and 139. The combination of systemic immunosuppression and PUVA therapy has been used to treat progressive diffuse morphea. 140
Linear scleroderma is a variant of localized scleroderma in which sclerotic areas of skin develop in a linear pattern.141. and 142. It is the second most common form of localized scleroderma after morphea. 143 It may occur on the head, trunk, or extremities; on the limbs it may extend the full length, leading to contractures of the joints that it crosses. 144 Muscle calcification is extremely rare. 145 A familial case has been recorded. 146
Linear scleroderma involving the frontoparietal area is referred to as en coup de sabre from its supposed likeness to the scar of a saber cut.147.148.149.150. and 151. Central nervous system and ophthalmic involvement are rare. 152 This variant, which is more likely to be bilateral than the other forms of linear scleroderma,153. and 154. may be associated with various degrees of facial hemiatrophy (Parry–Romberg syndrome).155.156. and 157. A study from Mexico City in 2002 found that it was possible to distinguish scleroderma en coup de sabre (SCS) from progressive facial hemiatrophy (Parry–Romberg syndrome). 158 Whereas cutaneous sclerosis was present in 8 of 13 patients with SCS, it was absent in all 9 patients with facial hemiatrophy. Cutaneous hyperpigmentation and alopecia were also more common in patients with SCS.158. and 159. The more frequent clinical features in facial hemiatrophy were total hemifacial involvement and ocular changes. 158 A retrospective study of 54 patients from the Mayo Clinic, reported in 2007, found 26 patients with SCS, 13 with Parry–Romberg syndrome, and 15 with both diseases. Bilateral disease was present in four patients. In addition to the association between this syndrome and SCS,160. and 161. other associations of Parry–Romberg syndrome have included borreliosis, 162 anti-double-stranded DNA antibodies, 163 and neurological involvement. 160 Rarely, linear scleroderma has been observed overlying the sclerosing bone dystrophy known as melorheostosis,164. and 165. or in association with hypertrichosis166. and 167. or systemic lupus erythematosus. 168 Osteolysis with subsequent fractures is a rare occurrence. 169 In many cases the lesions appear to have followed Blaschko’s lines.58.170.171. and 172. The simultaneous occurrence of linear scleroderma and homolateral segmental vitiligo has been reported. 173
The onset of linear scleroderma is sometimes abrupt and occasionally follows trauma. 141 Its mean duration is longer than that of plaque-type morphea and it is less likely to resolve as completely. Disease activity is mirrored by the titer of antihistone autoantibodies, which decrease with disease resolution. 174
Topical calcipotriol, combined with PUVA therapy, have been used to treat SCS. 151 UVA1 phototherapy can also be used for various sclerotic skin diseases. 175
Localized scleroderma is characterized by three outstanding features – the deposition of collagen in the dermis and subcutis, vascular changes, and an inflammatory cell infiltrate, particularly in early lesions. 176 These changes are now considered in detail. The epidermis may be normal, somewhat atrophic, or even slightly thicker than usual. 177
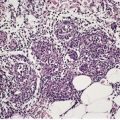
The dermis is increased in thickness and composed of broad sclerotic collagen bundles which stain strongly with the trichrome stain. Collagen also replaces the fat around the sweat glands and extends into the subcutis. In the latter site the collagen is homogenized and less compact than in the dermis (Fig. 11.1) and it shows only weak birefringence and trichrome staining; 178 there is an increased number of fibroblasts. However, the collagen in the subcutis stains strongly with the PAS stain in contrast to the very weak staining of that in the dermis. Mucopolysaccharides are present in the early lesions, particularly in the subcutis. Rarely, a secondary cutaneous mucinosis, with significant interstitial mucin, is present. In most cases of morphea the thickened collagen bundles are in the mid and deep reticular dermis.
Localized scleroderma (morphea). The recently deposited collagen stains weakly with the usual collagen stains and is devoid of elastic tissue. (Verhoeff–van Gieson)
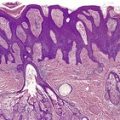
There is atrophy of adnexal structures, particularly the pilosebaceous units. Eccrine glands are situated at a relatively high level in the dermis as a result of the collagen deposited below them (Fig. 11.2). The arrectores pilorum are often hypertrophied. The mesenchymal elements of peripheral nerves are involved in the sclerotic process. 179
(A) Localized scleroderma (morphea). (B) Note the swollen collagen bundles, the atrophic sweat glands and the straight edge of the dermal–subcutaneous interface. (H & E)
The vascular changes are thickening of the walls of small blood vessels and narrowing of their lumen. In small arteries there is fibromucinous thickening of the intima.
The inflammatory cell infiltrate is composed of lymphocytes with some macrophages and plasma cells. It is distributed around blood vessels or more diffusely through the lower dermis and subcutis, particularly at the border of early lesions. The infiltrate is more marked in localized scleroderma than systemic scleroderma and in early rather than late lesions. The infiltrate is rarely heavy. 180 Immunohistochemical characterization of the infiltrate has shown the presence of T lymphocytes of both CD4 and CD8 subtype, as well as Langerhans cells and natural killer cells. 181
In guttate lesions the changes are more superficial with less collagen sclerosis but with subepidermal edema, resembling this feature of lichen sclerosus et atrophicus. Linear lesions may show a deeper and more diffuse inflammatory cell infiltrate extending into the underlying muscle. Vascular changes are usually prominent. Ossification of the dermis has been recorded. 182 The inflammation in the subcutis is also marked in the generalized form183 and in subcutaneous morphea; in both there may be marked fibrosis in the subcutis. 32
In subcutaneous morphea there is thickening and hyalinization of collagen in the deep dermis and in the septa and fascia.184.185. and 186. There is a mixed inflammatory cell infiltrate which includes some multinucleate giant cells. Lipomembranous (membranocystic) changes may be present. Some confusion has arisen because of the variable use of the term ‘subcutaneous morphea’. One group has suggested that the term ‘morphea profunda’ be used as an all-embracing one to include cases with dermal, subcutaneous and fascial involvement, while ‘subcutaneous morphea’ should be used for cases in which the subcutaneous fat is mainly affected. 184 According to this concept, eosinophilic fasciitis refers to the fascial component of morphea profunda. 184 The concept has not gained wide acceptance.
In keloidal nodules there are hyalinized thick collagen bundles associated with an increase in fibroblasts and mucin. 53Bullous lesions show subepidermal edema with dilated lymphatics in the underlying dermis.29.184. and 187. Erythrocytes are often present in the blister. 187
In superficial morphea the collagen deposition and inflammation are restricted to the superficial dermis.188. and 189. A mild lymphoplasmacytic infiltrate surrounds eccrine ducts in the superficial dermis. 63 There are no features of lichen sclerosus et atrophicus. Dermal elastic fibers are not appreciably diminished, but there is some loss of CD34-positive spindle cells. 188
The differentiation of morphea from the fibrotic lesions of acrodermatitis chronica atrophicans (see p. 579) can sometimes be difficult. 190 Although morphea and systemic scleroderma share many histological features, the lesions of morphea are usually more inflammatory than in systemic scleroderma. Furthermore, there may be some collagen deposition in the papillary dermis in morphea; 191 it is, of course, a feature of the superficial variant. In the study from Mexico City, referred to above, fibrosis was present in all cases with SCS, but only in two of nine cases with facial hemiatrophy. 158 Adnexal atrophy and mononuclear cell infiltrates were also more common in SCS cases. 158
Immunofluorescence is usually negative in the lesions of localized scleroderma although a few deposits of IgM may be found in the basement membrane zone and in small dermal blood vessels. 32 An increased number of cells in the dermis express factor XIIIa and vimentin, with a reduced number expressing CD34 in established lesions.192. and 193.
The cases reported as ‘self-involuting atrophoderma’ occurred as non-indurated, slightly depressed lesions on the lateral upper arm that disappeared spontaneously within a year. 194 There was some fibrosis of the lower dermis, but not the subcutis. This is best regarded as a variant of morphea.
Systemic scleroderma (systemic sclerosis) is an uncommon connective tissue disease characterized by symmetrical tightness, thickening and induration of the skin, Raynaud’s phenomenon, and sometimes involvement of one or more internal organs.197.198. and 199. The spectrum of systemic scleroderma ranges from a relatively mild form with limited acral skin involvement to a more rapidly progressive diffuse form with early and significant involvement of various internal organs.18. and 197.
The diffuse form accounts for 20–40% of cases of systemic scleroderma. There is usually truncal and acral skin involvement of abrupt onset, associated with the appearance of Raynaud’s phenomenon and constitutional symptoms. 197 Synovitis is common. Other features include esophageal hypomotility and strictures, rectal prolapse, sigmoid volvulus, nodular regenerative hyperplasia of the liver, primary biliary cirrhosis, idiopathic pulmonary fibrosis, pulmonary hypertension, Sjögren’s disease, thrombosis of major vessels,200. and 201. intrauterine fetal death, 202 and renal failure. 197 Neurotropic ulceration secondary to peripheral neuropathy is a rare occurrence. 203 Left ventricular non-compaction, myopathy, and polyneuropathy were associated with cutaneous sclerosis in one case. 204
The limited variant of systemic scleroderma typically affects older women. Raynaud’s phenomenon often precedes the onset of cutaneous thickening, which is usually limited to the digits. 205 Hair loss and anhidrosis are present in affected areas. Facial telangiectasia and cutaneous calcification often develop and there is an increased incidence of late-onset pulmonary hypertension. Anticentromere antibodies are present in up to 70–80% of patients.206.207.208.209.210. and 211.
Limited systemic scleroderma includes the condition referred to as the CREST syndrome,212. and 213. which derives its name from the clinical features of calcinosis, Raynaud’s phenomenon, esophageal dysfunction, sclerodactyly, and telangiectasia. 214 Not all of these features are invariably present, leading to suggestions that this term should be dropped in favor of the term ‘limited systemic scleroderma’. 20 Another variant combines Raynaud’s phenomenon, anticentromere antibodies, and digital necrosis without sclerodactyly. 215 Glomerulonephritis is a rare association. 216 A patient with localized systemic scleroderma developed abrupt thickening and induration of the skin following an episode of borreliosis. This component of her illness resolved after specific antibiotic therapy for the Borrelia infection. 217 Limited systemic sclerosis has been reported in association with discoid lupus erythematosus in two Japanese patients with anticentromere antibodies. 218
Pigmentary changes may be found in both the diffuse and the limited forms of systemic scleroderma. 219 They may take the form of vitiligo-like areas with perifollicular and sometimes supravascular sparing, diffuse hyperpigmentation with accentuation in sun-exposed areas, or pigmentary changes in areas of sclerosis.220. and 221. Reticulate hyperpigmentation is another variant. 222 Livedo reticularis and livedoid vasculitis with ulcers occur uncommonly. 201 Macrovascular involvement as detected by arteriography is not rare in patients with digital ulceration or gangrene. 223
Other clinical features of systemic scleroderma include a weak association with certain HLA antigen types, particularly DR5 and DR1, 224 and rare familial cases.225.226. and 227. Antinuclear antibodies are present in almost all patients, usually with a speckled or nucleolar pattern.208.228.229. and 230. Further characterization of these antibodies is possible now that specific nuclear macromolecules have been identified. 208 Autoantibodies to the Fc receptor are present in about 50% of cases, 231 while antibodies to Scl-70 (antitopoisomerase) are present in approximately 30% of patients.197.232. and 233. Patients with these ANA antibodies are more susceptible to vascular disease and pulmonary fibrosis. 234 A patient with systemic lupus erythematosus and topoisomerase I antibody has been reported. 235 A subset of patients with lupus-like features has anticardiolipin antibodies. 236 These antibodies may have a role in the genesis of vascular involvement related to systemic scleroderma. 237 Antibodies to the cytoplasmic antigen Ro/SSA are present in approximately one-third of cases. 238 Antineutrophil cytoplasmic antibodies of perinuclear type (p-ANCA) are present in a small number of patients. 239 Systemic angiitis has been reported as a rare complication of systemic sclerosis, but the p-ANCA status has been reported in only one of these cases.239. and 240. The serum levels of various enzymes and substrates involved in the sclerotic process have been used as an indicator of disease activity. They include xylosyltransferase (involved in proteoglycan metabolism),241. and 242. tissue inhibitors of metalloproteinase-2, 243 matrix metalloproteinase-9, 244 soluble vascular adhesion molecule 1 and E-selectin, 245 type I collagen degradation products, 246 and hyaluronan. 247
Although the pathogenetic basis for the fibrosis in scleroderma is still not elucidated, theories relate this to changes in the vascular system, immune disturbances, or alterations of fibroblast function.197.248.249.250. and 251. It is probable that these three factors will prove to be interdependent and interrelated. 250 Infection with parvovirus B19 has also been suggested as an etiological agent.252.253. and 254. Abnormal immune reactions to this virus might be a consequence of the infection. The virus is known to infect endothelial cells and stromal fibroblasts; it was found by PCR in these cells in all of the cases reported by Magro and colleagues. 253
Vascular changes include the formation of gaps between endothelial cells, alterations in the composition of type IV collagen in vascular basement membrane, 255 reduplication of the basal lamina, and disruption of endothelial cells. 249 It has been suggested that the endothelial cell is the principal target; 256 endothelial dysfunction precedes other cutaneous changes in systemic scleroderma.257. and 258. These changes are associated with increased serum levels of endothelial adhesion molecules and endothelium-associated cytokines. 259 An increase in the number of pericytes has also been observed in the marginal zones of active disease. 260 Cutaneous hypoxia, which results from the fibromucinous intimal change in larger vessels, may play a role in the modulation of dermal fibroblast activity. 261
Alterations in the immune system include the presence of autoantibodies and circulating immune complexes206. and 262. as well as an increase in the T-helper/T-suppressor cell (CD4/CD8) ratio.249. and 263. A clonal population of T cells has been found in lesional skin in patients with limited scleroderma; it is much less frequent in the diffuse form.264. and 265. Dominant T-cell clones have also been found in the peripheral blood. 266 There is also an association with other autoimmune diseases and a similarity to chronic graft-versus-host disease. It has been suggested that lymphokines and monokines produced by cells in the inflammatory infiltrate may play a role in fibroblast regulation;249.263. and 267. transforming growth factor-β (TGF-β) appears to be one of the most important (see below). Endothelin-1, derived from keratinocytes, appears to play an important role in the pathogenesis of the skin hyperpigmentation seen in patients with systemic scleroderma. 268 Monocytes may be responsible for the release of toxic free oxygen radicals, long thought to be involved in the pathogenesis of systemic sclerosis. 269 Their role in the pathogenesis of bleomycin-induced sclerosis is discussed elsewhere (see p. 313). Microchimerism has also been proposed as an initiating factor in the pathogenesis of scleroderma. It refers to persistence of low levels of fetal cells in a mother after childbirth. Such cells could provide a target antigen for both cell-mediated and humoral phenomena. 270
Numerous studies have attempted to elucidate the mechanisms controlling fibroblast activity in scleroderma. 249 There is an increase in the synthesis, deposition and degradation of collagen, proteoglycans, and fibronectin. The increased collagen may result from the accumulation of a distinct subpopulation of fibroblasts with an activated transcriptional level of collagen gene expression.271. and 272. As mentioned above, it seems that the increased fibroblast activity in scleroderma results from the activity of cytokines, the most important of which are interleukin-4 (IL-4) 273 and TGF-β.274. and 275. This growth factor can, in turn, induce the production of three interrelated substances – connective tissue growth factor (CTGF), platelet-derived growth factor (PDGF), and tissue inhibitor of metalloproteinase-3 (TIMP-3) 276– which influence the mitogenic activity and function of fibroblasts resulting in increased collagen production.272. and 277. Whereas TGF-β is responsible for the early collagen deposition, it is maintained by the subsequent activity of CTGF. 278 CTGF is particularly involved in the regulation of type I collagen production. 278 Decorin overexpression reduces TGF-β levels. 117 Collagen deposition may also be enhanced by the release of cytokines and polyamines from epidermal keratinocytes. 279 Lysyl oxidase, which initiates crosslinkage of collagen and elastin, is increased in amount. 280 Furthermore, the increased deposition of collagen is enhanced by reduced collagenase activity.281. and 282. This deposition is accompanied by a significantly increased amount of collagen crosslink pyridinoline. 283 It is possible that endothelial damage followed by platelet aggregation initiates the release of PDGF and the subsequent cascade of events. 284 Some of the type III collagen that is initially produced retains the aminopeptide on its surface, resulting in the formation of thin collagen fibrils, 30–40 nm in diameter.285. and 286. These fibrils may form bundles in the subcutis or at the advancing edge or be mixed with larger diameter fibrils. 285 In the later stages, the ratio of type III to type I collagen is normal. 287 Reactive oxygen species (ROS) produced by fibroblasts may also contribute to the pathogenesis of scleroderma. 288
A review of the treatment options for scleroderma, published in 2002, listed the following therapeutic options: (1) vasodilators; (2) immunosuppressant drugs, such as methotrexate, cyclosporine (ciclosporin), cyclophosphamide, and extracorporeal photopheresis; and (3) antifibrotic agents, such as D-penicillamine, colchicine, interferon gamma, and relaxin. 137 In a recent study, D-penicillamine therapy resulted in a reduction in skin involvement and improvement of other organ systems. 289 Hemopoietic stem cell transplantation is another treatment that has been trialed in severe cases. 290 A patient with therapy-recalcitrant generalized bullous scleroderma was successfully treated by extracorporeal photopheresis and mycophenolate mofetil. 291 The tyrosine kinase inhibitor imatinib has also been trialed in this condition. 292
In limited systemic scleroderma, the use of the 5-phosphodiesterase inhibitor sildenafil citrate (Viagra) has produced improvements in both the Raynaud’s phenomenon and digital dexterity. 293 The endothelin receptor antagonist bosentan has been used to treat the digital ulcers in this form of the disease.294. and 295. Erythropoietin has also been used for this purpose. 296
The histopathological changes in systemic scleroderma are similar to those described above in the localized forms, although minor differences exist (Fig. 11.3). The inflammatory changes are less marked in systemic lesions and the deposition of collagen can be quite subtle in the early stages, particularly on the fingers. Edema fluid is present in the papillary and reticular dermis of early lesions. 300 Vascular changes are sometimes more prominent, particularly severe intimal fibrosis in small arteries and arterioles (Fig. 11.4). 301 Dermal telangiectasia is common in the limited form. 302 These vessels may show evidence of recent or old thrombosis and adventitial fibrosis. 200 Sclerosis of the papillary dermis can be seen in morphea but it is absent in systemic scleroderma. 191
Systemic scleroderma with thick collagen bundles in the dermis. Inflammation is absent. (H & E)
Systemic scleroderma. There is fibromuscular thickening of the wall of a small artery with luminal narrowing. (H & E)
Other changes described include calcification, 303 an increase in mast cells in the dermis of early lesions300. and 304. and of clinically uninvolved skin, 305 and pigmentary changes corresponding to the clinical changes. For example, the vitiligo-like areas show an absence of melanocytes and of melanin in the basal layer.219. and 220. Amyloid is a rare finding. 306Digital lesions with the histology of focal mucinosis have been described. 307
Direct immunofluorescence is usually negative, although a few cases have been described with a speckled nuclear pattern in epidermal cells similar to that seen in mixed connective tissue disease (see below).308. and 309. Immunoperoxidase techniques show a reduced number of CD34-positive cells. 310
Mixed connective tissue disease (MCTD) is a distinct clinical syndrome sharing some clinical features of systemic lupus erythematosus, scleroderma, and polymyositis. 313 It is associated with the presence of circulating antibody to ribonucleoprotein.314. and 315. Antibodies to the Sm antigen and to native DNA are usually absent. 316 Swollen or sclerotic fingers, Raynaud’s phenomenon, and arthritis are important clinical features, leading to a suggestion that the acronym SRA is more appropriate than MCTD. 317 Less constant clinical features include muscle tenderness and proximal weakness, lymphadenopathy, alopecia, esophageal hypomotility, and pigmentary disturbances.317.318. and 319. Approximately 20% develop restrictive lung disease. 317
MCTD usually runs a chronic and benign course and shows a good response to systemic corticosteroids.
The terms scleromyositis and sclerodermatomyositis have been used for cases with overlap features of scleroderma and dermatomyositis.320. and 321. The sera of these patients often show a homogeneous nucleolar pattern. 230 PM-SCL autoantibodies are present in about 70–90% of cases. 322 Other features of this disease include HLA-type associations, eczema of the hands (‘mechanic’s hands’), and interstitial lung disease.322. and 323. Autoimmune hepatitis and sarcoidosis were present in one case. 324
If lupus-like lesions are present clinically, then a biopsy from such an area will show the features of cutaneous lupus erythematosus. Even in lesions that are not clinically typical of lupus erythematosus, the histological changes may resemble subacute lupus erythematosus. 325 In the early stages, a biopsy from a swollen finger reveals marked dermal edema with separation of collagen bundles. 318 In later lesions, dermal sclerosis resembling that seen in scleroderma may be present. The walls of vessels in the subcutis may be thickened with luminal narrowing.
Direct immunofluorescence of uninvolved skin shows a characteristic pattern of speckled epidermal nuclear staining, with specificity for IgG. 326
In eosinophilic fasciitis (Shulman’s syndrome) there is a sudden onset, sometimes following strenuous physical activity, of symmetrical induration of the skin and subcutaneous tissues of the limbs.327. and 328. There is usually sparing of the fingers. Localized variants, with involvement of part of a limb, have been reported. 329 The disease usually begins in mid-adult life, but no age is exempt.330. and 331. Other clinical features include peripheral eosinophilia332. and 333. and hypergammaglobulinemia, 328 although rare cases with specific immunoglobulin deficiencies have been reported. 334 Serum levels of TIMP-1 (tissue inhibitor of metalloproteinase) are elevated. 335 Visceral involvement and Raynaud’s phenomenon are usually absent. 336 An association with cutaneous T-cell lymphoma has been reported.337. and 338. The majority of affected patients experience a complete or near complete recovery after 2–4 years, usually following steroid or PUVA-bath therapy but sometimes occurring spontaneously.339. and 340.
Eosinophilic fasciitis is regarded as a variant of scleroderma.341.342. and 343. As in scleroderma, fibroblasts in the involved skin of patients with eosinophilic fasciitis exhibit an activated phenotype. 344 Progression to scleroderma has been documented in several circumstances, 345 including a group of patients with the Spanish toxic oil syndrome (see p. 313). Patchy lesions of morphea are sometimes present on the trunk, further evidence of the close association between eosinophilic fasciitis and the various scleroderma syndromes.330. and 346. Eosinophilic fasciitis has also been reported as a manifestation of chronic graft-versus-host disease, sometimes in association with lichen sclerosus. 347 It may also develop as a paraneoplastic phenomenon. 348 The spirochete Borrelia burgdorferi has been implicated in the etiology of some cases of eosinophilic fasciitis,349. and 350. but specifically excluded in other cases. 351 Similar changes have also resulted from exposure to trichloroethylene, 352 and from radiation, 353 the subcutaneous injection of phytonadione (phytomenadione) in the treatment of hypoprothrombinemia354 and the ingestion of products containing L-tryptophan. A virtual epidemic of the eosinophilia-myalgia syndrome occurred in the USA and Japan in 1989 following alterations in the manufacturing techniques of L-tryptophan. This condition is likely to become of historical interest only.355.356.357.358.359. and 360. Atorvastatin has been implicated in one case of eosinophilic fasciitis. 361
Although spontaneous recovery is possible, most patients respond to corticosteroids. 362 Various other therapies have been used as no single treatment works in all cases. 363 Dapsone is sometimes effective. 363
The earliest changes occur in the interlobular fibrous septa of the subcutis and the deep fascia. There is edema and an infiltration of lymphocytes, histiocytes, plasma cells, and eosinophils. 364 Eosinophils are sometimes quite prominent, but in most instances there are only focal collections of these cells. 365 Lymphoid nodules may also be present.
Eventually, there is striking thickening of the deep fascia and septa of the subcutis with fibrosis and hyalinization of the collagen.364. and 366. This process extends into the deep dermis where there is atrophy of appendages associated with the sclerosis of the lower dermis. Inflammatory changes may also extend into the fibrous septa of the underlying muscle, 367 but this process is not the same as eosinophilic myositis/perimyositis. 368 Similar changes were seen in the L-tryptophan-related cases369 although there was greater dermal involvement in this condition; mucin and dermal sclerosis were often seen.370.371. and 372.
Immunoglobulins and C3 have been present in the walls of vessels in the fascia and subcutis in some cases.
Atrophoderma (of Pasini and Pierini) is an uncommon yet distinctive form of dermal atrophy consisting of one or more sharply demarcated, depressed, and pigmented patches.375.376.377. and 378. The color varies from bluish to slate gray or brown. 379 There is no induration or wrinkling. 379 Individual lesions are round or ovoid and may coalesce to give large patches of involvement. There is a predilection for the trunk, particularly the back. 376 In one report, the lesions had a zosteriform distribution. 380 The onset, which is insidious, usually occurs in adolescence. 375 It has been reported in three groups of siblings.381.382. and 383. Lesions may slowly progress over many years or they may persist unchanged.
Atrophoderma is regarded by some as an abortive variant of morphea,384. and 385. an opinion favored by some overlapping clinical and histopathological features and by isolated reports of progression to either morphea or systemic sclerosis. 386 It is probably the same condition as superficial morphea. 65 Ackerman believes it is morphea, unqualified. 387 Others regard it as a distinct disease entity on the basis of the dermal atrophy, the usual absence of sclerosis, 375 and the unique glycosaminoglycan metabolism. 388 Unfortunately, this controversy is based on a small spectrum of clinical experience. 389
The etiology and pathogenesis are unknown, but it has been suggested that macrophages and T lymphocytes that are present around the vessels in the dermis may play some role. 384 In one report, 10 of the 26 patients studied had elevated serum antibodies to Borrelia burgdorferi. 376 Twenty of the 25 patients treated with antibiotics showed clinical improvement. 376 Another patient has shown a dramatic response to hydroxychloroquine. 390
There is dermal atrophy but this may not be apparent unless adjacent normal skin is included in the biopsy for comparison (Fig. 11.5).379. and 391. The collagen bundles in the mid and deep dermis are sometimes edematous or slightly homogenized in appearance.375. and 385. Elastic fibers are usually normal, although there may be some clumping and loss of fibers in the deep dermis. 375 Adnexal structures are usually preserved. There is a perivascular infiltrate of lymphocytes and a few macrophages; rarely, plasma cells are prominent. 376 The infiltrate is usually mild in the upper dermis and somewhat heavier around vessels in the deep dermis. Some superficial vessels may be mildly dilated.
Atrophoderma. There is some thinning of the dermis (on the back). Collagen bundles are slightly thickened and homogenized. (H & E)
The epidermis is usually normal, apart from hyperpigmentation of the basal layer. There may be a few melanophages in the superficial dermis.
In the one study known to the author, the collagen and elastic fibers were normal. 384
The sclerodermoid disorders are a heterogeneous group of diseases in which lesions develop that may mimic clinically and/or histopathologically the changes found in scleroderma.17.392. and 393. The sclerodermoid disorders, some of which are discussed elsewhere, as indicated below, include:
• sclerodermoid graft-versus-host disease
• stiff-skin syndrome
• Winchester syndrome
• GEMSS syndrome
• pachydermoperiostosis
• pachydermodactyly
• acro-osteolysis
• chemical- and drug-related disorders
• paraneoplastic pseudoscleroderma
• nephrogenic systemic fibrosis
• lichen sclerosus et atrophicus
• post-stripping cutaneous sclerosis
• scleredema (p. 359)
• scleromyxedema (p. 355)
• porphyria cutanea tarda (p. 497)
• chronic graft-versus-host disease (p. 312)
• chronic radiation dermatitis (p. 528)
• Werner’s syndrome (p. 328)
• progeria (p. 329).
No detailed mention need be made of the carcinoid syndrome394 and of phenylketonuria,395. and 396. both of which are exceedingly rare causes of sclerodermatous skin lesions. A spectrum of sclerodermoid disorders has been reported in phenylketonuria including morphea, atrophoderma of Pasini and Pierini, and lichen sclerosus et atrophicus.397. and 398. Melorheostosis (OMIM 155950), a rare sclerosing bone dysplasia, is sometimes associated with skin changes overlying the bone changes. They include scleroderma and focal skin thickening. 399 In one patient increased procollagen α1(I) mRNA expression was found in dermal fibroblasts but no obvious dermal thickening was present. 400 Sclerodermoid changes have also followed the cutaneous eruption of rhabdomyolysis, 401 and following the formation of an arteriovenous fistula for hemodialysis. 402
It should be noted that squeezing the skin with forceps, during a biopsy procedure, can produce an artifactual change locally in the dermis which resembles scleroderma somewhat on histopathological examination. Separation of the collagen bundles at the margins of this zone or artifactual changes in the overlying epidermis may also be present.
Scleroderma-like lesions may develop in chronic graft-versus-host disease which, by definition, occurs more than 100 days after transplant (see p. 55). Most patients who develop this uncommon complication of GVHD have disseminated sclerosis of the trunk and the proximal extremities. 403 Atrophy of the skin is associated with a severe clinical evolution. Sometimes only localized or bullous lesions are present.404.405. and 406. Koebnerization has been reported in one case. 407 Lichen sclerosus, eosinophilic fasciitis, and discoid lupus erythematosus have all been reported as manifestations of chronic graft-versus-host disease.347.405.408. and 409. Most cases develop in adults; pediatric cases are rare.410. and 411.
Antinucleolar, anticentromere, and anti-Scl-70 antibodies are usually not present in patients with sclerodermatous GVHD. 403
The disease is usually quite refractory to standard therapies including immunosuppression and extracorporeal photopheresis. 412 Extracorporeal photopheresis seems to be less efficacious than previously thought. 413 Therapy with UVA1 has proved satisfactory. 412 Imatinib is another potential treatment. 414 Ultrasound can be used to monitor response to therapy. 415
The sclerodermatous lesions of chronic GVHD show similar features to scleroderma, although the papillary and upper reticular dermis are involved at an earlier stage and there may be extension of the fibrosis into the subcutis. 416 The lichenoid changes of GVHD may also be present. 403 It may occur without a preceding lichenoid stage. 413 Secondary mucinosis is a rare finding. 417 In early lesions, the histological changes can be quite subtle despite the clinical features being quite overt.
The stiff-skin syndrome (OMIM 184900) is characterized by stony-hard skin, particularly on the buttocks and thighs, mild hypertrichosis, and limitation of joint mobility.418.419.420. and 421. It differs from restrictive dermopathy (see p. 324), but has similarities to congenital fascial dystrophy.422. and 423. Both appear to be genetically determined abnormalities of dermal and fascial collagen. Abortive forms have been described. 424
The Parana hard-skin syndrome (OMIM 260530) was described in a Brazilian family. It differs from the stiff-skin syndrome by severe growth retardation and a more malignant course.
In the cases reported as stiff-skin syndrome there have been mild fibrosis of the dermis and subcutis, but no inflammation. 419 An increase in dermal mucin was noted in some of the earlier cases but this is not a consistent feature. 425 In cases reported as fascial dystrophy, the deep fascia is thickened 4–6-fold. 422 Again, there is no inflammation. 422
The Winchester syndrome (OMIM 277950) is an exceedingly rare inherited disorder of connective tissue which consists of dwarfism, carpal-tarsal osteolysis, rheumatoid-like small joint destruction, and corneal opacities.426. and 427. Cutaneous manifestations include a thick, leathery skin with areas of hypertrichosis and hyperpigmentation. 428 A case in which the skin thickening was symmetrically banded has been reported. 429
The Winchester syndrome is caused by a mutation in the gene encoding matrix metalloproteinase-2 (MMP2) which maps to chromosome 16q13. 430 Winchester and NAO (nodulosis–arthropathy–osteolysis) syndromes are allelic. MMP2 is also known as type IV collagenase as it specifically cleaves type IV collagen.
One study reported an abnormal oligosaccharide in the urine of two unrelated patients with this condition. 431 It is possible that this and another case431. and 432. are examples of infantile systemic hyalinosis (see p. 387) and not the Winchester syndrome.
There is increased pigmentation of the basal layer and some thickening of the dermis. Fibroblasts are markedly increased in number, although in late lesions there are only a few fibroblasts in the thickened masses of amorphous collagen. 427 A perivascular lymphocytic infiltrate is also present. There is no increase in mucopolysaccharides in the dermis.
GEMSS syndrome (OMIM 137765), a rare, autosomal dominant disorder, features glaucoma, lens ectopia, microspherophakia, stiffness of the joints and shortness. Sclerosis of the skin is sometimes present. This appears to be due to enhanced gene expression of TGF-β1. 433 The gene defect has not yet been characterized.
The changes resemble those seen in systemic scleroderma.
The clinical manifestations of this rare syndrome (OMIM 167100) include digital clubbing, thickening of the legs and forearms resulting primarily from periosteal new bone formation at the distal ends of the long bones, and progressive coarsening of facial features with deeply furrowed, thickened skin on the cheeks, forehead, and scalp (cutis verticis gyrata).434. and 435. Pachydermoperiostosis has an insidious onset, usually in adolescence, and a self-limited course. There is a predilection for males. 436 A familial incidence is sometimes present and in these cases the inheritance is thought to be autosomal dominant with incomplete penetrance and variable expressivity of the gene.437. and 438. No candidate gene has yet been identified.
Associated clinical conditions have included myelofibrosis, 439 protein-losing enteropathy, 440 and psoriatic onychopathy. 441
This condition has also been referred to as primary hypertrophic osteoarthropathy to distinguish it from a secondary form which is usually associated with an intrathoracic neoplasm.442.443.444. and 445. Facial and scalp changes, usually less severe than in pachydermoperiostosis, have been reported in some individuals with the secondary form.434.446.447. and 448. Finger clubbing may be the sole manifestation in some relatives of patients with pachydermoperiostosis, indicating the overlap between these conditions.434.442. and 449.
The pathogenesis is unknown, although many mechanisms have been postulated for the secondary form. Increased peripheral blood flow is found in secondary hypertrophic osteoarthropathy, but not in the primary variant. 450 One study has shown an increased tissue sensitivity to circulating sex hormones, and this could induce enhanced tissue epidermal growth factor/TGF-α production and utilization. 451 Another study has found that fibroblasts from affected skin synthesize decreased amounts of collagen but increased amounts of the small dermatan sulfate-containing proteoglycan decorin. 452
The epidermis may be normal or mildly acanthotic. There is a diffuse thickening of the dermis with closely packed, broad collagen bundles. Some hyalinization of the collagen is usually present. Fibroblasts are increased in number in some areas. The subcutis may also participate in the fibrosing reaction. Elastic fibers are usually normal, although variations have been recorded. Acid mucopolysaccharides are sometimes increased in the dermis. 438 In late stages there is some thickening of capillary walls with an increase in pericapillary collagen. 450
Other changes include a variable, usually mild, perivascular and periappendageal chronic inflammatory cell infiltrate and prominence of sebaceous and eccrine glands. 454
Pachydermodactyly is characterized by fibrous thickening of the lateral aspects of the proximal interphalangeal joints of the fingers.455.456.457.458.459. and 460. In contrast, knuckle pads, which have similar histological appearances, involve the dorsal aspect of the finger joints.
Pachydermodactyly and knuckle pads are usually regarded as localized forms of superficial fibromatosis. Accordingly, they are considered with the fibromatoses in Chapter 34, pages 816, 817.
Acro-osteolysis refers to lytic changes in the distal phalanges. There is a familial form, an idiopathic form with onset in early adult life, and an occupational variant related to exposure to vinyl chloride. 461 Cutaneous lesions have been described in only some idiopathic cases; 461 in contrast, they are a characteristic feature of occupational acro-osteolysis. 462 There are sclerodermoid plaques on the hands accompanied by Raynaud’s phenomenon. 392 With altered work practices, occupational acro-osteolysis should become a historical disease. 463
The dermis is thickened, with swollen collagen bundles and decreased cellularity. There is usually no significant inflammation and there is no calcinosis. Elastic fibers are often fragmented.
Sclerodermoid lesions may develop in the skin following occupational exposure to polyvinyl chloride (acro-osteolysis, see above), vinyl chloride monomer, 464 trichlorethylene, 465 perchlorethylene, aromatic hydrocarbon solvents, herbicides, 466 certain epoxy resins, and silica.463.467.468.469.470. and 471. Silica has also been implicated in the etiology of scleroderma itself.467. and 472. In recent years, there has been considerable controversy about the relationship between scleroderma and other connective tissue diseases and the use of breast prostheses containing silicone gel.470.473.474.475. and 476. Notwithstanding this debate about the possible systemic effects of extravasated silicone, it is acknowledged that a localized, dense collagenous reaction may ensue following such an event (see p. 394).
The injection of phytonadione (phytomenadione, vitamin K1),477.478.479.480. and 481. polyvinylpyrrolidone (a former plasma expander), 482 or pentazocine17. and 483. will result in a localized sclerodermatous reaction. The injection of narcotic drugs may result in the ‘puffy hand syndrome’, a form of lymphedema that mimics edematous scleroderma. 484
The ingestion of an olive oil substitute – rapeseed oil, denatured with aniline – produced a multisystem disease of epidemic proportions in Spain some years ago. 485 Sclerodermoid lesions developed in the skin. 486 A similar multisystem illness followed the ingestion of products containing L-tryptophan following alterations in the manufacture of this product in 1989. 355 In both conditions, the tissue fibrosis may result from the stimulation of fibroblasts by cytokines such as TGF-β and platelet-derived growth factor. 355
The chemotherapeutic agent bleomycin will produce cutaneous sclerosis, particularly involving the fingers, in addition to its other complications of alopecia, cutaneous pigmentation, and pulmonary toxicity.487.488. and 489. A sclerodermoid reaction has also been produced by peplomycin, an analogue of bleomycin. 490 The cutaneous lesions produced by bleomycin in particular, and to a lesser extent by some of the other agents listed above, are self-limiting, with some resolution of the lesions after withdrawal of the offending agent. Experimentally, mice with bleomycin-induced dermal sclerosis had a reduction in this sclerosis following injection of superoxide dismutase, supporting a role for superoxide radicals in the pathogenesis of the fibrosis. 491 A sclerodermoid reaction has been produced by recombinant interleukin-2 (aldesleukin) used in the treatment of renal carcinoma. 492 The second-generation anticancer drug uracil-tegafur (UFT) has been associated with a sclerodermatous reaction. 493 A similar reaction is also produced by docetaxel and paclitaxel, two of the newer antineoplastic drugs belonging to the group of taxanes.494.495.496.497. and 498. Gemcitabine, a new nucleoside analogue, used in the treatment of certain solid cancers, 499 and methysergide, used in migraine prophylaxis, have both produced sclerodermoid changes of the lower extremities. 500 Sclerodactyly-like changes have been produced by capecitabine, a drug that is metabolized into 5-fluorouracil. 501 The sclerodermoid reaction produced by doxorubicin and cyclophosphamide in a patient with breast cancer involved 80% of total body area. 502
The changes resemble quite closely those found in systemic scleroderma. In bleomycin-induced lesions, the homogenized collagen is often most prominent around blood vessels and adnexal structures.
Sclerotic skin lesions resembling systemic scleroderma are a rare complication of malignancies. This paraneoplastic syndrome is most often seen with lung cancer, plasmacytoma, and carcinoids.503. and 504. Marked expression of α1(I)-collagen and connective tissue growth factor (CTGF) mRNA, but not TGF-β1, was found in fibroblasts. 503
The changes in the dermis resemble those seen in systemic scleroderma. The fibrosis sometimes extends into the subcutis. 504
Although considered in this chapter in a previous edition, nephrogenic systemic fibrosis (nephrogenic fibrosing dermopathy) is histologically similar to scleromyxedema, although it does have changes in the subcutaneous fat not seen in scleromyxedema. Accordingly, it is considered in Chapter 13, page 358.
Lichen sclerosus et atrophicus (LSA) is a chronic disorder with a predilection for the anogenital region of middle-aged and elderly women.505.506.507.508. and 509. Patients present with itching, pain, and/or dyspareunia. 510 Cases in childhood are uncommon.511.512.513. and 514. They may be mistaken for child sexual abuse. 515 About 20% of the patients have extragenital lesions, these sometimes occurring without coexisting genital involvement. 516 Extragenital sites which are affected include the upper part of the trunk, the neck, the upper part of the arms, the flexor surfaces of the wrists, and the forehead. Very rarely, palmar, 517 plantar518. and 519. and digital520 skin, the face,521.522. and 523. scalp,524. and 525. lip,526. and 527. mouth528 and even a surgical529 or burn scar,530. and 531. chronic wound, 532 stoma,533. and 534. and a vaccination site535 have been involved. Nail dystrophy is rare. 536 Extragenital lesions may rarely follow Blaschko’s lines.537. and 538. Extragenital lesions are very rarely photoaggravated. 539
LSA may involve the glans, prepuce, or external urethral meatus of uncircumcized prepubertal or adolescent males, resulting in phimosis.540.541.542.543.544. and 545. These lesions, also known in the past as balanitis xerotica obliterans, are not associated with extragenital involvement, 540 although isolated extragenital lesions may be seen in other males.546. and 547. LSA is the usual cause of secondary phimosis in prepubescent boys. 548 LSA has developed in a case of male-to-female gender reassignment in skin formerly from the scrotum. 549 HPV is present in a significant number of penile lesions in children.550. and 551. A case has followed the use of alprostadil as an intracavernous injection for penile dysfunction. 552 Thyroid disease and psoriasis are commonly associated conditions. 510 LSA is exceedingly rare in chronic graft-versus-host disease. 347
LSA commences as flat, ivory to white papules that coalesce to form plaques of varying size and shape. These develop follicular plugging and progressive atrophy leading to a parchment-like, wrinkled, flat or slightly depressed scar (‘cigarette paper atrophy’). Vulval lesions may have secondary lichenification from the pruritus-related scratching or they may coexist with hypertrophic areas, the so-called ‘mixed vulval dystrophy’. 506 Infrequently, hemorrhagic bullae form553.554.555.556.557.558. and 559. and these may be complicated by the subsequent development of milia. 560 Small nodules and keratotic papules have been recorded as an unusual clinical manifestation.561. and 562. Linear lesions have also been described. 522 Pigmentation due to massive melanin incontinence is another rare finding. 563 As already mentioned, lesions in LSA are leukodermic. This may come about by decreased melanin production, a block in transfer of melanosomes to keratinocytes, and melanocyte loss. 564
Usually, the disorder is slowly progressive with periods of quiescence. Spontaneous involution may occur, particularly in girls565 at or about the menarche.566.567.568.569. and 570.
There is controversy concerning the relationship of LSA to morphea.571. and 572. Ackerman believes that LSA is a superficial variant of morphea. Although many authors have reported small numbers of cases of LSA coexisting with or superimposed upon morphea,67. and 573. it is suggested, but not universally accepted, 574 that these patients have morphea with secondary lymphedema and sclerosis of the superficial dermis mimicking LSA both clinically and pathologically. 572 In most, but not all instances, there have been no genital lesions. 575 Some patients with LSA have had coexisting autoimmune diseases.576.577.578. and 579. Other rare associations include glucose intolerance or diabetes mellitus, 580 vitiligo, 581 and sclerodermatous GVHD. 582 It has developed after allogeneic stem cell transplantation. 583 The role of HPV in LSA is controversial, despite its reported role in LSA-related squamous cell carcinoma of the penis (see below),410.584. and 585. and in penile LSA. 586 HPV has also been found in penile LSA in children (see above).
Extragenital lesions never undergo malignant degeneration, although in the genital region there may, uncommonly, be coexisting or subsequent squamous cell carcinoma.508.587.588.589. and 590. In these circumstances, the tumor usually arises in the hyperplastic areas of what is a mixed vulval dystrophy. 568 Interestingly, there is increased p53 but not Ki67 expression in vulval lesions of lichen sclerosus et atrophicus when compared to non-vulval lesional skin.591. and 592. The p53 changes may be of etiological significance in the development of some squamous cell carcinomas of the vulva arising in LSA. 593 Although epigenetic inactivation of p16INK4a occurs as an early event, it is insufficient for malignant transformation. 594 Malignant change has been recorded in up to 5.8% of penile LSA.595.596. and 597. Most of these cases had concomitant HPV infection, 595 although a subsequent study has refuted this association. 598 Of 20 patients with squamous cell carcinoma (SCC) of the penis, 11 had a clinical history and/or histological evidence of LSA. 599 In a larger series of 207 cases of SCC of the penis, 68 patients were identified with LSA. 600 When LSA was associated with malignancy it was often associated with low-grade squamous intraepithelial lesions. 600
Although the etiology of LSA is unknown, attention has been directed at the role of Borrelia burgdorferi, which has been detected by a modified silver stain and immunoperoxidase techniques in lesional skin.84.85. and 86. It has also been demonstrated by PCR-based techniques, focus-floating microscopy, 601 and serology. Most of the studies have been from Austria or nearby European countries.602. and 603. Some Borrelia-associated cases have been reported from Japan. 604 Attempts at detecting this organism in UK, US, and Australian cases have been unsuccessful.97.604. and 605. It is possible that this geographical association is related to the presence of the genotypes B. garinii and B. afzelii in Europe, but not in the USA, where B. burgdorferi sensu stricto is the usual species of Borrelia found. This particular strain does not appear to be associated with LSA. B. burgdorferi can also be detected in cases of morphea, Lyme disease, and atrophoderma of Pasini and Pierini; this latter condition has been reported in patients with LSA. 606 A patient with hepatitis C infection who developed lichen sclerosus–lichen planus overlap has been reported. 607
In LSA there are numerous epidermotropic and dermal lymphocytes that are CD8+, CD57+. This profile is usually associated with viral diseases, autoimmune diseases, and malignancies. 608 Morphea also exhibits CD57+ lymphocytes. Clonally expanded populations of T cells have been reported in the infiltrate. The low percentage of clonal T-cell receptor-γ DNA argues against a neoplastic disease, but rather for a local immune disorder, probably against an antigen of infectious origin.609.610. and 611. Immunological changes appear to occur at all levels of the skin. 612 Circulating basement membrane zone antibodies have been found in a small number of patients with LSA of the vulva. 613 Their presence did not correlate with any clinical feature. 613
The histological changes suggest that significant alteration of the extracellular matrix is occurring.614. and 615. This may, in part, be mediated by the decreased epidermal expression of CD44, which can produce increased hyaluronate accumulation in the superficial dermis. 616 Increased levels of the extracellular hyaluronic acid (HA)-binding protein ITI (inter-α-trypsin inhibitor) is closely implicated in the accumulation of HA in the broad hyalinized zone of the superficial dermis. 617 Circulating IgG autoantibodies to extracellular matrix protein 1 have been found in about 75% of patients with LSA. 618
Another finding in LSA is a loss of androgen receptors in lesional skin with disease progression. 619 This may be a secondary effect rather than of etiological significance. Estrogen receptor expression is increased in the vulva in LSA; it may be implicated in the etiopathology of the disease. 620
Certain HLA types (particularly DQ7 but also DQ8 and DQ9) are more frequent in patients with LSA.579.621. and 622. Familial cases are rare. 623
Treatment of LSA is usually with a topical ultrapotent corticosteroid, such as clobetasol propionate ointment 0.5% for a limited time.624.625.626. and 627. In one series of 327 patients with vulvar LSA, topical ultrapotent steroids gave symptom relief in most, and completely reversed the skin changes in about one-fifth of patients. 626 Another series found improvement but not cure in elderly patients with vulvar LSA. 625 In younger patients who achieved complete remission, 50% had relapsed after 16 months of cessation of the treatment. 625 Treatment may possibly prevent the development of vulvar squamous cell carcinoma. 625 Other treatments used have included acitretin (for lesions on the scalp), 525 methotrexate, 628 tacrolimus ointment, 624 low-dose ultraviolet A1 (UVA1) phototherapy, 629 psoralen-UVA (PUVA) therapy, 630 and narrowband UVB phototherapy. 631 The use of tacrolimus for genital disease has been criticized because of its potential for producing squamous cell carcinoma. 632 All three studies using ultraviolet therapy were for extragenital lesions only.629.630. and 631. Therapeutic strategies using antioxidants have been suggested on the basis of one study that showed oxidative damage of lipids, DNA, and proteins in LSA. 633
Established lesions show hyperkeratosis, follicular plugging, thinning of the epidermis, and vacuolar alteration of the basal layer (Fig. 11.6). There is a broad zone of subepidermal edema with homogenization of collagen and poor staining in hematoxylin and eosin preparations. In later lesions, this zone becomes more sclerotic in appearance and shows more eosinophilia. Basement membrane thickening also occurs. 635 Expression of collagen IV and VII is increased. 636 There is dilatation of thin-walled vessels in the zone and sometimes hemorrhage. Beneath the edema there is a diffuse, perivascular infiltrate of lymphocytes, predominantly of T-cell type in the mid dermis. This infiltrate is sometimes quite sparse in established vulval lesions and it may contain a few plasma cells and histiocytes. Mast cells and liberated mast cell granules are also present. 605 In vulval lesions there is also more diversity of epidermal changes, with hyperplastic areas in mixed dystrophies. 637 Vulvar LSA without associated carcinoma has a mean epidermal thickness more than three times that of extragenital LSA. It resembles lichen simplex chronicus. LSA adjacent to carcinoma tends to show exaggerated epidermal thickening, basal atypia, and loss of the edematous-hyaline layer. 638 The appendages are usually preserved.
(A) Lichen sclerosus et atrophicus. (B) There is orthokeratotic hyperkeratosis, some basal vacuolar change and subepidermal edema and homogenization of collagen. In established lesions the infiltrate is deeper and more dispersed. (H & E)
A spectrum of vascular changes occurs in LSA ranging from a rare leukocytoclastic vasculitis, to a not uncommon lymphocytic vasculitis, to an exceedingly rare granulomatous phlebitis.639. and 640. Three forms of lymphocytic vasculitis have been recorded: (i) concentric lymphohistiocytic infiltrates with lamination of the adventitia by basement membrane material, seen typically in penile lesions; (ii) lymphocytic vasculitis with dense perivascular lymphocytic cuffing with occasional fibrin deposition in vessel walls and subendothelial infiltration by lymphocytes (Fig. 11.7); and (iii) intramural lymphocytic infiltrates in large muscular vessels. 640
Lichen sclerosus et atrophicus. A lymphocytic vasculitis is present. (H & E)
In the early stages, elastic fibers are pushed downwards by the edematous zone and subsequently destroyed. 641 Fibrillin is reduced in the upper dermis, but it is normal immediately beneath the basement membrane. 642 In contrast, elastic fibers are normal or increased in morphea. 643 Small amounts of acid mucopolysaccharide may be found in this zone. The basement membrane may focally fragment and PAS-positive material may be found in the subjacent dermis, partially as homogeneous clumps. 641 Numerous invaginations and holes are present in the basement membrane zone (BMZ) at the level of the lamina lucida and lamina densa; in contrast, the continuity of the BMZ is preserved in morphea.644. and 645. In bullous lesions of LSA, the split occurs below the lamina densa. 645
In early lesions, the inflammatory infiltrate is quite heavy and is superficial and band-like, mimicking lichen planus (Fig. 11.8). Basal apoptosis and vacuolar change (Fig. 11.9) accompany the infiltrate; that is, there are features of the lichenoid tissue reaction (interface dermatitis). Overlap syndromes with lichen planus have also been suggested.646. and 647. Features favoring a diagnosis of LSA over lichen planus include basilar epidermotropism, basement membrane thickening, epidermal atrophy, loss of papillary dermal elastic fibers, paucity of cytoid bodies, and a lack of wedge-shaped hypergranulosis.648. and 649. As the edematous zone broadens, the infiltrate is pushed downwards and becomes more dispersed and usually less intense. Changes also involve adnexal structures. 650
Lichen sclerosus et atrophicus. Early lesion with a heavy lymphocytic infiltrate in the upper dermis. (H & E)
Lichen sclerosus et atrophicus. Interface changes with (A) basal cell death and (B) vacuolar change. (H & E)
Lesions of LSA may also mimic mycosis fungoides because of the presence of epidermotropism and focally coarse collagen bundles in the papillary dermis. Monoclonal T lymphocytes have also been recorded. 651Incidentally, mycosis fungoides may present clinically with lesions resembling LSA. 652
Melanocytic proliferations developing in LSA may show atypical features.653. and 654. Melanocytic nevi often resemble persistent melanocytic nevi. Melanocytes, nevoid or malignant, proliferating contiguously with fibrotic or sclerotic collagen, contain abundant melanin, diffusely express HMB-45, and have a higher Ki-67 labeling index than ordinary melanocytic nevi. 653
In presumptive cases with coexisting morphea, the absence of vacuolar alteration, the lack of a well-defined inflammatory infiltrate beneath the thickened dermis, and the presence of deep dermal changes of morphea are features supporting a diagnosis of morphea without coexisting LSA. 572
Microscopic features of LSA have been recorded, as an incidental finding, in acrochordons655 and in the skin tag/folds of the perineum known as infantile pyramidal (perineal) protrusion. 656 Another study of this protrusion showed no histological evidence of LSA. 657
The term ‘post-stripping cutaneous sclerosis’ is preferred to the rather cumbersome title of the original report – ‘post-stripping sclerodermiform dermatitis’. 660 The condition presents as multiple hypopigmented and indurated plaques distributed in a linear arrangement along the path of a previously stripped saphenous vein. 660
There is diffuse dermal sclerosis, sometimes extending into the subcutis. There is superficial telangiectasia and/or lymphangiectasia with mild epidermal atrophy. There is often a mild deep perivascular infiltrate of lymphocytes but this was not present in two cases seen by the author. Inflammation may not be present in older lesions.
Collagen is increased in several conditions, but not necessarily in the manner seen in scleroderma and the sclerodermoid disorders. Connective tissue nevi and hypertrophic scars and keloids have been arbitrarily included in this section; they could also be regarded as tumor-like proliferations of fibrous tissue, other examples of which are discussed in Chapter 34.
The dermis is usually thickened and somewhat sclerotic in lipodermatosclerosis. It has been considered with the panniculitides (see Ch. 17, p. 472).
Connective tissue nevi are cutaneous hamartomas in which one of the components of the extracellular connective tissue – collagen, elastic fibers, or glycosaminoglycans – is present in abnormal amounts. 661 They can be subclassified on the basis of the component predominantly involved:
• collagen type
collagenoma
shagreen patch
• elastin type
elastoma
• proteoglycan type
nodules in Hunter’s syndrome.
Sometimes there are alterations in more than one component of connective tissue, and these lesions may simply be categorized as connective tissue nevi.662. and 663. Only the connective tissue nevi of collagen type will be discussed in this section.
Collagenomas (connective tissue nevi of collagen type) are rare hamartomas of the skin in which there is an increase in dermal collagen. They usually present as asymptomatic, firm, flesh-colored plaques and nodules, 0.5–5.0 cm in diameter, on the trunk and upper part of the arms. 661 The ear, 664 sole of the foot,665. and 666. and vulva667 are rare sites of involvement. There may be several lesions668 or up to a hundred or more, with an onset in adolescence.669. and 670. Rapidly growing (eruptive) collagenomas have been reported in the multiple endocrine neoplasia type I syndrome, 671 and in pregnancy. 672 Eruptive collagenomas have also been reported as the only lesion.673. and 674. Uncommonly, they occur in a zosteriform or linear distribution.675.676.677.678.679.680. and 681. A family history is sometimes present (familial cutaneous collagenoma) and in these cases there is autosomal dominant inheritance.669.682.683.684. and 685. Familial cutaneous collagenomas (OMIM 115250) result from a mutation in the LEMD3 gene, as seen in the Buschke–Ollendorff syndrome.686. and 687.
Associated clinical features include a cardiomyopathy, 661 Down syndrome,688.689. and 690. and occult spinal dysraphism. 691 Uncommonly, the connective tissue nevi associated with the Buschke–Ollendorff syndrome (see p. 334) are of collagenous composition rather than of the usual elastic tissue type.692.693.694. and 695. A connective tissue nevus of collagen type has been reported in association with pseudo-Hurler polydystrophy (mucolipidosis III), 696 and in Proteus syndrome, in which the nevi are often acral, sometimes of the plantar cerebriform type.697.698. and 699. Plantar collagenomas can occur in the absence of the Proteus syndrome. 700
Solitary collagenomas are sometimes quite large, as seen with the cerebriform or ‘paving stone’ variants on the sole of the foot.701. and 702. Sometimes a connective tissue nevus is associated with cutis verticis gyrata, a descriptive term for a condition of the scalp in which deep furrows and convolutions are present. The folds of skin may, however, have normal morphology.703. and 704. Other pathological associations of cutis verticis gyrata include lymphedema, 705 mental retardation, 706 adipocyte proliferation, acromegaly, 707 misuse of anabolic substances, 708 myxedema, Noonan syndrome,706. and 709. tumors, and the insulin resistance syndrome. 710 Cutis gyrata, usually on the scalp, can be seen in the Beare–Stevenson syndrome (OMIM 123790) characterized by mutations in the transmembrane region of fibroblast growth factor receptor (FGFR)-2 located at chromosome 10q26. Acanthosis nigricans, skin tags, and anogenital anomalies are other features of this syndrome. 711 There is a reduction in expression of tuberin in connective tissue nevi associated with the tuberous sclerosis complex. 712
Collagenomas must be distinguished from sclerotic fibromas (see p. 815) which present as tumor-like nodules. They have a characteristic histological appearance with dense collagen bundles, often in a storiform arrangement. Athlete’s nodules are related to, but different from, collagenomas. One such example is the dermal nodule found in the sacrococcygeal region of bicycle riders. 713
The epidermis is usually normal, although an overlying epidermal nevus has been reported. 714 There is thickening of the dermis, sometimes with partial replacement of the subcutis. The collagen bundles are broad and have a haphazard arrangement. 715 Elastic fibers are more widely spaced, but this may represent a dilution phenomenon. 715 Sometimes the elastic fibers are thin and fragmented. Some of these cases may represent papular elastorrhexis (see p. 344). 673 There is no increase in mucopolysaccharides. Dermal dendrocytes are reduced using antibodies against factor XIIIa. 716 Calcification was present in one reported case. 717
The shagreen patch is a distinct clinical variant of collagenoma, found exclusively in those with tuberous sclerosis. It consists of a slightly elevated, flesh-colored plaque of variable size, usually on the lower part of the trunk. 719 It has the appearance of untanned leather. Smaller ‘goose flesh’ papules may form as satellite lesions.
There are dense sclerotic bundles of collagen, with an interwoven pattern, in the reticular dermis. 720 Fibroblasts appear hypertrophied. 720 There is no inflammatory infiltrate or increase in vascularity. The overlying epidermis is usually flat, although sometimes it has the pattern of acanthosis nigricans. 719
White fibrous papulosis consists of multiple, pale, discrete, non-follicular lesions on the lateral and posterior neck.721.722.723. and 724. The lesions are asymptomatic and gradually increase in number; eventually there may be 10–100 papules, measuring 1–3 mm in diameter. There is some clinical resemblance to pseudoxanthoma elasticum, but there are no angioid streaks. Most of the reported cases have been from Japan and Italy. 725
There is significant overlap between cases reported as white fibrous papulosis of the neck and those reported as papillary-dermal elastolysis (acquired elastolysis of the papillary dermis simulating pseudoxanthoma elasticum) – see page 348. Accordingly, it has been suggested that both entities be combined under the term ‘fibroelastolytic papulosis of the neck’. 725
The etiology is unknown, but in view of the onset late in life, the condition may be the result of an age-related change in dermal collagen.722. and 726. Cultured fibroblasts express increased amounts of collagen type I mRNA. 727
White fibrous papulosis has some resemblance to a connective tissue nevus. There is a circumscribed area of thickened collagen bundles, involving the papillary and mid dermis. 722 Elastic fibers are usually reduced in areas of fibrosis. 725
Hypertrophic scars and keloids are a variation of the optimal wound healing process.728.729.730. and 731. Keloids have been defined as cutaneous scars that extend beyond the confines of the original wound, and hypertrophic scars as raised scars that remain within the boundaries of the wound. 732 As the clinical distinction may be blurred, the usefulness of these definitions has been questioned. 733 Equally, the histopathological definitions proposed below have been criticized on the grounds that they do not always correlate with the above clinical definitions. 728 The differences between keloids, hyperplastic scars, and the optimal (normal) wound healing process are of degree only, and it is not surprising that cases with overlap features exist. Keloids may be quite disfiguring and have therefore attracted much more attention than hypertrophic scars.
Keloids are firm, variably pruritic masses, usually at the site of injury. 734 They may be unevenly contoured. Early lesions are erythematous, while older lesions are usually pale, although occasionally they may be pigmented. 733 Sites of predilection are the upper part of the back, the deltoid and presternal areas, and the ear lobes. Rare sites include the genitalia, eyelids, and even palms and soles. 733 They usually develop over a period of weeks or months. Attempted surgical excision of a keloid results in regrowth of a larger lesion unless some concurrent effective therapeutic measures, such as radiation therapy or the use of cultured epithelial autografts, are adopted.733.735.736.737. and 738. Recurrence is significantly less frequent in hypertrophic scars.735. and 739.
Factors leading to the formation of keloids and hypertrophic scars include race, increased skin tension in a wound, age of the patient, wound infection, site, as already mentioned, the use of isotretinoin or cyclosporine therapy, and a predisposition to scar hypertrophy.728.732.740.741. and 742. Keloids may develop within adolescent striae, 743 and secondary to therapeutic cupping. 744 Atrophic scars have followed acupuncture. 745 Hypertrophic scars have also followed patch testing, BCG vaccination, and carbon dioxide laser ablation of plantar warts in cyclosporine-treated patients.746.747. and 748. Keloidal plaques have been reported on the lower extremities in a patient with Ehlers–Danlos syndrome type IV. 749 Keloids are more common in black races. 750 They have a predilection for individuals under 30 years of age. A study of 14 pedigrees with familial keloids showed an autosomal dominant mode of inheritance with incomplete clinical penetrance and variable expression. 751 Nine up-regulated genes have been identified in keloid tissue. These genes, especially NNP-1 (novel nuclear protein-1), probably contribute either directly or indirectly to keloid formation. 752
Numerous experimental studies have been designed to identify the etiology and pathogenesis of keloids. Fibroblasts isolated from keloids often synthesize normal amounts of collagen, 753 although proline hydroxylase activity, a marker of collagen synthesis, is higher in keloids than in hypertrophic scars or normal wounds. 754 A lower rate of apoptosis and of mutations in p53 occurs in keloid fibroblasts.755. and 756. Other studies suggest an important role for various cytokines, particularly TGF-β which is a potent chemotactic factor for fibroblasts757.758.759.760. and 761. and stimulates them to produce major matrix components including collagen. 757 Heat shock protein 47 (HSP47) is overexpressed in keloid fibroblasts and is another mechanism involved in collagen accumulation. 762 Interleukin-15 (IL-15) also appears to be involved. 763 The relative amounts of type III collagen are increased. There is also increased expression of type I and III collagen mRNA.761.764. and 765. There is a major suppression of pro-α1(I) type I collagen gene expression in the dermis after excision of a keloid followed immediately by intrawound injection of triamcinolone acetonide, an explanation for the beneficial action of corticosteroid injections. 766 Expression of the gli-1 oncogene is strongly elevated in keloids compared with scars. 767 Cultured keloid fibroblasts demonstrate the bioenergetics of cancer cells. 768 Collagenase activity may be normal or increased in keloids; 769 decreased levels have been found in hypertrophic scars.770. and 771. Activation of matrix metalloproteinase-9 has been demonstrated during in-vitro mechanical compression in hypertrophic scars. It could be the effector mechanism for the regression that results from mechanical compression. 772 Changes in the extracellular matrix proteins occur in keloids. 773 Their interaction with TGF-β may explain their mode of action.774. and 775. Tenascin C, biglycan, integrins, and decorin are increased in some hypertrophic scars and keloids.776.777.778. and 779. Expression of dermatopontin (a multifunctional protein of the extracellular matrix which influences collagen assembly), is decreased. 780 Angiotensin-converting enzyme (ACE) is significantly higher in pathological scar tissue than in normal and wounded skin. 781 Vascular endothelial growth factor of epidermal origin appears to be increased in keloids.782. and 783. The abnormal cutaneous sensations that occur in hypertrophic scars may be the result of up-regulation of opioid receptors in lesional skin. 784
It appears that collagen accumulates in keloids and hypertrophic scars because there are more fibroblasts present, making more collagen than in normal wound healing, and this collagen may be protected from degradation by proteoglycan and specific protease inhibitors.733. and 785. Some degradation of collagen does occur as there is increased prolidase activity, 786 but the collagen synthesis exceeds any breakdown that occurs.
Many different therapies have been used in the treatment and prevention of hypertrophic scars and keloids, an indication that no one treatment ‘cures all’. Topical treatments used include pressure therapy, 787 silicone gel sheeting and ointment, 788 polyurethane dressing, onion extract, imiquimod 5% cream, and vitamins A and E. 789 Intralesional corticosteroids, laser therapy, 790 cryosurgery, tissue-engineered allografts, 791 intralesional 5-fluorouracil,792. and 793. or interferon-α2b, 794 and dermojet injections of bleomycin795 have also been used. Asiaticoside, a derivative of the plant Centella asiatica (Indian pennywort), has been used for the treatment of hypertrophic scar for many years, as an ingredient of Gotu Kola, a traditional herbal medicine. 796 It appears to enhance the expression of Smad 7, which has an inhibitory effect on scar fibroblasts. 796 Recombinant adenovirus-mediated double suicide gene therapy has been used experimentally to destroy keloid fibroblasts. 797
Early keloids show abundant fibrillary collagen (Fig. 11.10). Mature keloids have a characteristic appearance with broad, homogeneous, brightly eosinophilic collagen bundles in haphazard array (Fig. 11.11). Fibroblasts are increased and are found along the collagen bundles with an orientation similar to the accompanying collagen. The AgNOR count of the fibroblasts is significantly increased. 799 There is also abundant mucopolysaccharide, particularly chondroitin-4-sulfate, between the bundles. Keloids have reduced vascularity when compared with hypertrophic scars and normal healing wounds. 800 Keloids are usually elevated above the surrounding skin surface. The overlying epidermis may be thin and beneath it there are often some telangiectatic vessels. A sparse, chronic inflammatory cell infiltrate may surround these peripheral vessels. A study from Taiwan in 2004 reported that in scars with no detectable keloidal collagen, the feature(s) favoring the diagnosis of keloid were: non-flattened epidermis, non-fibrotic papillary dermis, a tongue-like advancing edge, a horizontal cellular fibrous band in the upper reticular dermis, and a prominent fascia-like band. 801 Not all these features mirror what is described in this section.
An early keloid with thick hyaline collagen replacing the scar tissue. (H & E)
(A) Established keloid. (B) The thick hyaline bundles of collagen contrast with the more normal ones below. (H & E)
In contrast, hypertrophic scars are only slightly elevated, if at all, above the surrounding skin. The collagen bundles are characteristically oriented parallel to the skin surface, as are the accompanying fibroblasts (Fig. 11.12). They are markedly increased in number, although there is some reduction in number with time. Capillaries are generally oriented perpendicular to the skin surface and these may be surrounded by a sparse inflammatory cell infiltrate within the scar itself. There is little mucin except in early lesions. Elastic tissue is sparse or absent. Subepidermal clefting sometimes develops overlying a scar; 802 bullae are rare. 803
Hypertrophic scar. There are parallel bundles of cellular collagen contrasting with those of the dermis below the scar. (H & E)
Mast cells are increased in both hypertrophic scars and keloids. Dystrophic calcification and bone may occasionally develop, particularly in abdominal scars. 804
Attention has been drawn recently to the presence of S100-positive cells, including spindle cells with mild atypia, in cutaneous scars. 805 Care needs to be taken in these circumstances to avoid overdiagnosis of desmoplastic melanoma. Conversely, desmoplastic melanomas can also be misdiagnosed as scars. Alpha-smooth muscle actin has been found in both hypertrophic scars (70%) and keloids (45%). 801 CD34 and factor XIIIa are not expressed in scars.806. and 807.
Keloids exhibit numerous fibroblasts with prominent Golgi complexes and abundant rough endoplasmic reticulum. 808 Myofibroblasts have usually not been demonstrated, 808 although it has been speculated that they may be present in early lesions. 809 Very fine elastic fibers can be seen in hypertrophic scars on electron microscopy, although they cannot be demonstrated by light microscopy. 810
Striae distensae are a common finding in adolescents of both sexes,811. and 812. particularly females, and in pregnancy (striae gravidarum).813.814. and 815. They were found in 83% of Korean students aged 15 to 17. 816 In another study 87% of primigravid pregnant women developed striae gravidarum. 817 Risk factors for the development of striae gravidarum include a history of breast or thigh striae, a family history, and the race of the patient. 818 Striae distensae are also found following prolonged heavy lifting.819. and 820. Striae form in association with the excess corticosteroid of Cushing’s disease, with systemic steroid therapy, and with prolonged topical use of steroid preparations.821. and 822. They develop on the abdomen, lower part of the back, buttocks, thighs, and female breasts. They are significantly more common on the thighs of girls and on the knees of boys.816. and 823. They were confined to the site of a navel piercing in one pregnant patient. 824 Striae may also occur in HIV-positive persons receiving protease inhibitors, such as indinavir. 825 Ulceration of striae has been reported in a patient with systemic lupus erythematosus. 826
Striae show a progression of clinical appearances, commencing as flat, pink lesions which broaden and lengthen and assume a violaceous color. 821 The terms striae caerulae (blue) and striae nigrae (black) have been used to describe striae distensae that appear darker than usual due to increased melanization. 827 The color gradually fades to leave a white, depressed scar. The direction of the striae is conditioned by the mechanical forces responsible, but they are usually linear. They have been attributed to the stretching of corticosteroid-conditioned skin. 819 It is possible that only minimal stress is required in some circumstances, such as adolescence. 828 This continuous strain on the dermal extracellular matrix may remodel the elastic fiber network in susceptible individuals. 829 The proportion of crosslinked to unlinked collagen may also be of critical pathogenetic importance. 828
Striae distensae are basically scars. The epidermis is flat with loss of the normal rete ridge pattern. The dermis may be thinned, suggesting that this condition would be better considered with the atrophic collagenoses. 833 Dermal collagen bundles are arranged in parallel array, resembling a scar. 830 There is an increase in glycosaminoglycan content in striae. 829 The Verhoeff–van Gieson stain for elastic tissue will usually show a reduction in elastic fibers, but the orcein and Luna stains will often show many additional fine fibers. 831 Using specific markers, it has been shown that the numbers of vertical fibrillin fibers beneath the dermal–epidermal junction and elastin fibers in the papillary dermis are significantly reduced. The elastin and fibrillin fibers in the deep dermis show realignment parallel to the skin surface. 829 In older lesions, thicker elastic fibers can be seen in the affected skin. 832 Early lesions (striae rubrae) are rarely biopsied. They have been reported to show vascular dilatation and a mild perivascular inflammatory cell infiltrate. Another study has suggested that the early stages consist of mast cell degranulation, followed by an influx of activated macrophages that envelop fragmented elastic fibers. 834 The increase in elastic fibers noted in advanced lesions is presumably a later stage of repair.
Only a few cases of fibroblastic rheumatism have been described since the initial report in 1980.835.836.837.838.839. and 840. There is a sudden onset of symmetrical polyarthritis and Raynaud’s phenomenon and the development of cutaneous nodules, 0.2–2 cm in diameter, which are found mainly on the hands. They may be more extensive. 841 A patient with cutaneous nodules, but without polyarthritis, has been reported. 842 The cutaneous lesions resolve spontaneously after several months. 840 The nature of the disease is unknown. The suggestion that it is a variant of non-Langerhans cell histiocytosis seems unlikely. 843 Nevertheless, it does mimic multicentric reticulohistiocytosis clinically. 844
The cutaneous nodules show a proliferation of plump spindle cells, as well as fibroblasts, set in a background of thickened collagen bundles having a whorled pattern. 837 The plump cells are myofibroblasts. 838 There is some loss of elastic tissue, but there is preservation of the vasculature. A mild, perivascular, chronic inflammatory cell infiltrate is sometimes present.
Collagenosis nuchae (nuchal fibroma) presents clinically with diffuse induration and swelling, usually of the back of the neck, accompanied by features suggesting low-grade inflammation.845.846.847. and 848. Extranuchal involvement has been recorded. 848 There is a male predominance. Non-destructive recurrences occasionally develop after excision. 848 Collagenosis nuchae has been reported in association with scleredema, 849 diabetes, 848 lipoma and traumatic neuroma, 850 and Gardner’s syndrome. 851 In Gardner’s syndrome, the lesions may occur in multiple sites and in unusual locations. 852 The term ‘Gardner-associated fibroma’ has been suggested for these lesions. 852
Computed tomography has been used to visualize the lesions. 853
Thick, disorganized collagen bundles partly replace the subcutaneous fat. The collagen merges almost imperceptibly with the lower dermis and the ligamentum nuchae. There are no inflammatory cells present, despite the clinical impression of inflammation (Fig. 11.13). Furthermore, there are very few fibroblasts, distinguishing the lesion from a fibromatosis. However, there has been a report of a desmoid fibromatosis having areas resembling collagenosis nuchae. 854 Many of the cells express CD34; a few contain factor XIIIa. 851 There is no increase in mucin, as seen in scleredema (see p. 359). Nerve entrapment is a frequent occurrence. 846 The architectural features of nuchal fibroma are subtly different from the formless sheets of collagen without neuromatosis change seen in the Gardner-associated fibroma. 855
Collagenosis nuchae (nuchal fibroma). Fibrous tissue replaces the subcutaneous fat. There is no inflammation. (H & E)
Lipodermatosclerosis is characterized by circumscribed, indurated inflammatory plaques on the lower extremities. Sometimes the plaques are quite large. The erythema and woody induration are often misdiagnosed as cellulitis. The condition is mentioned here because of the dermal thickening and fibrosis. There is considerable involvement of the subcutis, and this entity is therefore considered in more detail with the panniculitides (see Ch. 17, p. 472).
Weathering nodules are asymptomatic, white or skin-colored nodules measuring 2–3 mm in diameter. They have a gritty texture. 856 The lesions are usually bilateral and multiple with small chains of lesions producing a scalloped appearance of the helix. Reported cases have all been in elderly males with an outdoor occupation or hobby. 856 The lesions appear to be more common than is currently recognized.
The lesions are composed of a spur of fibrous tissue in which there is a focus of metaplastic cartilage which is sometimes in continuity with the cartilage of the ear. The fibrous tissue sometimes extends to the undersurface of the epidermis. It is relatively acellular but it may contain spindle-shaped cells that are negative for S100 protein and factor XIIIa. They differ from elastotic nodules of the ear which have severe actinic elastosis, not cartilaginous metaplasia with a spur of fibrous tissue as seen in weathering nodules. 857
The microscopical assessment of dermal atrophy can be difficult in the early stages. It requires a knowledge of the regional variability in skin thickness. The age of the patient is also relevant, since there is some atrophy of the skin in the elderly. Technical artifacts in the preparation of histological sections can also influence dermal thickness.
The following conditions, discussed below, can be associated with a decrease in dermal thickness:
• aplasia cutis congenita
• focal dermal hypoplasia
• focal facial dermal dysplasia
• pseudoainhum constricting bands
• keratosis pilaris atrophicans
• corticosteroid atrophy
• linear atrophoderma of Moulin
• acrodermatitis chronica atrophicans
• restrictive dermopathy.
In addition, dermal thinning can be seen in type IV Ehlers–Danlos syndrome (see p. 327) and also in some forms of Marfan’s syndrome (see p. 350). The dermis is usually thinned in striae distensae (see p. 319) but, because of the scar-like features, this condition has been considered with the hypertrophic collagenoses. The unique case with membranocystic degeneration of collagen fibers, associated with some deposition of fat, and with the clinical appearance of xanthomatosis, defies classification. 858
Aplasia cutis congenita is the term applied to a heterogeneous group of disorders in which localized or widespread areas of skin are absent at birth. Its incidence is 1–3 per 10 000 births. 859 The defect is most often limited to the vertex of the scalp,860. and 861. but other parts of the body such as the trunk or limbs may be affected, often symmetrically, with or without accompanying scalp lesions.862.863.864.865. and 866. Scalp defects range in size from 0.5 to 3 cm in diameter but larger defects up to 10 cm in diameter have been recorded.867. and 868. Other recorded associations include limb defects,869.870.871. and 872. mental retardation, 873 split cord malformations, 874 oculoectodermal dysplasia, encephalocraniocutaneous lipomatosis,875. and 876. epidermal and organoid nevi,877.878. and 879. epidermolysis bullosa,859.880.881.882.883. and 884. chromosomal abnormalities, 863 fetus papyraceus,877.885. and 886. and focal dermal hypoplasia.863.873. and 887. The Adams–Oliver syndrome (OMIM 100300) refers to the combination of aplasia cutis congenita and transverse limb defects. 888 Other systemic abnormalities, including coarctation of the aorta, may be present.889. and 890. The gene responsible has not been identified.
The MIDAS (microphthalmia, dermal aplasia sclerocornea) syndrome is another distinct variant of aplasia cutis congenita presenting as linear facial skin defects. The Xp deletion syndrome, MLS (microphthalmia with linear skin defects) syndrome (OMIM 309801), and Gazali–Temple syndrome are similar conditions. 891 It is lethal in males. There is usually a microdeletion at Xp22, 892 resulting in defects in the HCCS (human holocytochrome C synthase) gene.893. and 894. The gene for focal dermal hypoplasia is nearby (see below). This syndrome is distinct from focal dermal hypoplasia, but it could be included with focal facial dermal dysplasia (see below).
Two publications have attempted to define distinct clinical subtypes, based on the location of the skin defects and the presence of associated malformations (Table 11.1).863. and 877. Some cases have an autosomal dominant inheritance with reduced penetrance of the gene; 895 others appear to be the result of gene mutation.863. and 896. The condition has been reported in one of monozygotic twins. 897 Other factors that may be etiologically significant in individual cases include amniotic adhesions, intrauterine trauma, drugs, particularly the antithyroid drug methimazole (thiamazole),898.899. and 900. biomechanical forces from the hemispheric growth of the brain, 901 and ischemia resulting from placental infarcts902 or associated with the condition fetus papyraceus.877.885. and 903. A case mimicking aplasia cutis congenita, but resulting from a congenital nevus, has been reported. 904
The defect in the skin presents as an ulcer, a membranous or bullous lesion, or an area of atrophic scarring. 905 Sometimes there is a ring of coarse, long hair surrounding the scalp defect (the ‘hair collar sign’).906.907. and 908. Dermal melanocytosis was present in one such case. 909 Lesions on the scalp usually heal with cicatricial alopecia. 910 An abnormal tendency to cutaneous scarring has been reported in two siblings with this condition. 911
Skin dimpling, which may be confused with aplasia cutis congenita, has been reported as a complication of second trimester amniocentesis. 912
The epidermis may be absent or thin, with only two or three layers of flattened cells. The underlying dermis is usually thin and composed of loosely arranged connective tissue in which there is some disarray of collagen fibers (Fig. 11.14). 913 The dermis may resemble a scar. 914 Elastic fibers may be reduced, increased, or fragmented. 915 Appendages are absent or rudimentary. 916 The subcutis is usually thin.
Aplasia cutis congenita of the scalp. Appendages are lost and the dermis is composed of thin, widely-spaced bundles of collagen. (H & E)
Focal dermal hypoplasia (Goltz or Goltz–Gorlin syndrome – OMIM 305600) is a rare syndrome with multiple congenital malformations of mesoderm and ectoderm, affecting particularly the skin, bones, eyes, and teeth.917.918.919.920. and 921. It is caused by mutations in the gene encoding the human homologue of Drosophila melanogaster Porcupine (PORCN), an endoplasmic reticulum protein involved in the secretion of Wnt proteins, which are important for the development of affected organs in focal dermal hypoplasia.922.923. and 924. The gene maps to Xp11.23. It is inherited as an X-linked dominant trait, which usually is lethal in the male: 925 however, this is not absolute as some male cases have been reported.926.927. and 928. Father-to-daughter transmission is a rare event. 929
Cutaneous manifestations include widespread areas of dermal thinning in a reticular, cribriform and linear pattern, soft yellowish nodules representing either herniations of subcutaneous fat through an underdeveloped dermis or heterotopic fat (a fat nevus),930. and 931. and linear or reticular areas of hyper- or hypopigmentation, often following Blaschko’s lines.917.932.933. and 934. Lentigo-like pigmented macules at the periphery of atrophic lesions have been reported. 935 Focal loss of hair, nail changes, total absence of skin from various sites, exophytic granulation tissue, 936 apocrine hidrocystomas, 927 lichenoid hyperkeratotic papules, giant papillomas, 937 and periorificial tag-like lesions may also occur.917.932. and 938. Skeletal abnormalities include syndactyly, polydactyly, and longitudinal striations of the metaphyseal region of long bones (osteopathia striata).933.939.940.941. and 942. Dermal hypoplasia may be a manifestation of the Proteus syndrome. 943 Rarely, only cutaneous abnormalities are present in focal dermal hypoplasia.
Experimental studies using fibroblasts from affected skin have shown that synthesis of collagen by individual fibroblasts is normal, but that there is an abnormality in cell kinetics with reduced proliferative activity of fibroblasts. 944 An absence of collagen type IV from the basement membrane zone has been reported.927. and 945.
The epidermis is usually normal. There is a marked reduction in the thickness of the dermis, with some thin, loosely arranged collagen fibers in the papillary dermis. Adipose tissue continuous with the subcutaneous fat extends almost to the undersurface of the epidermis in some areas. Clusters of adipocytes may be anywhere in the dermis or in a perivascular location. 946 The extreme degree of attenuation of the collagen seen in focal dermal hypoplasia is not present in nevus lipomatosus, in which mature fat also replaces part of the dermis; furthermore, the clinical presentations of the two conditions are quite distinct.
Inflammatory cells, sometimes quite numerous, have been present in biopsies of focal dermal hypoplasia taken in the neonatal period.925. and 947. Other findings have included the presence of increased numbers of blood vessels and of some elastic tissue within the fat, a feature not present in normal subcutaneous fat. 948 Elastic fibers are markedly diminished in the dermis. 949
One study has shown fine filamentous tropocollagen within and between collagen bundles. 950 Another has found loosely arranged collagen bundles composed of few fibrils scattered in the extracellular matrix. 949 Rough endoplasmic reticulum and Golgi complexes are not prominent in dermal fibroblasts, which are smaller and diminished in number.949. and 950. Elastic fibers are scarce and of normal morphology. Multilocular fat cells, which are regarded as young fat cells, are often seen. 950 Disruption of the basement membrane zone is also present. 945
Focal facial dermal dysplasia (Brauer syndrome – OMIM 136500) and facial ectodermal dysplasia (Setleis syndrome – OMIM 227260) are rare genodermatoses that are now thought to be the same condition. 951 They are characterized by congenital, usually symmetrical, scar-like lesions of the temple.952.953. and 954. Lesions may extend onto the face. There is often a spectrum of associated facial anomalies. 952 The tetralogy of Fallot was present in one case. 955 Inheritance is usually as an autosomal dominant trait with incomplete penetrance; 956 other patterns of inheritance have been reported in what are probably clinical variants. 957 A ‘hair collar’ may surround the defect.958. and 959. This entity has been regarded in the past as a variant of aplasia cutis congenita915 or as an ectodermal dysplasia. 960
A specific variant of facial lesion that indicates the close relationship between focal facial dermal dysplasia and aplasia cutis congenita is the preauricular skin defect characterized by oval, atrophic patches distributed in a linear pattern on the preauricular region of the face. 961 It is thought to be the result of a persistent ectodermal groove in the region of the fusion between the maxillary and mandibular facial prominences. 961
The term ‘pseudoainhum’ refers to congenital constriction bands which may take the form of shallow depressions in the skin or deep constrictions associated with gross deformity or even amputation of a limb or digit.962. and 963. The term ‘ainhum’ is a West African name for similar constrictive lesions, usually of the fifth toe, that lead eventually to spontaneous amputation and that probably result from the effects of repeated trauma in the genetically predisposed African patients. The amniotic band syndrome is a similar condition. 964 Closely related are the raised linear bands that developed in the first few months of life in a few patients with localized constriction. In one case the lesions formed at the sock line, 965 although this may be an unrelated condition.
Acquired lesions developing later in life may occur in association with scleroderma, psoriasis, syringomyelia, leprosy, and pachyonychia congenita.966. and 967.
There is marked thinning of the dermis with finger-like projections of fibrous tissue extending into the underlying subcutis. Elastic tissue may be increased in this region.
The term ‘keratosis pilaris atrophicans’ refers to a group of three related disorders in which keratosis pilaris is associated with mild perifollicular inflammation and subsequent atrophy, particularly involving the face. Differences in the location and the degree of atrophy have been used to categorize these three conditions – keratosis pilaris atrophicans faciei (ulerythema ophryogenes), keratosis follicularis spinulosa decalvans and atrophoderma vermiculata (folliculitis ulerythematosa reticulata). All three conditions are regarded as congenital follicular dystrophies with abnormal keratinization of the superficial part of the follicles, and are therefore considered with other follicular abnormalities in Chapter 15, page 433. Dermal atrophy is an inconstant and disputed feature.
In all three variants of keratosis pilaris atrophicans there is follicular hyperkeratosis with atrophy of the underlying follicle and sebaceous gland. There is variable perifollicular fibrosis which may extend into the surrounding reticular dermis as horizontal lamellar fibrosis. The dermis may be reduced in thickness.
Cutaneous atrophy is an important complication of the long-term topical application of corticosteroids, especially if occlusive dressings are used or the steroids are fluorinated preparations.968.969. and 970. The injection of corticosteroids into the skin will also produce local atrophy;971. and 972. rarely, atrophic linear streaks develop along the lines of the overlying lymphatic vessels draining the injection site.973.974.975. and 976. Telangiectasia and striae are other complications that may develop.977. and 978.
Profound digital collagen atrophy has been reported in a patient with Cushing’s syndrome. 979 Elevated plasma cortisol levels were present secondary to an adrenal adenoma.
The pathogenesis of corticosteroid atrophy is not completely understood. It appears to involve diminished synthesis and enhanced degradation of collagen.980.981. and 982.
Epidermal thinning with loss of the rete ridges is an early change. The superficial dermis often has a loose texture and there may be telangiectasia of superficial vessels. The reticular dermis is reduced in thickness only after prolonged topical therapy. If atrophy is present the collagen bundles may appear thin and lightly stained. 984 At other times the collagen appears homogenized. 985 In some areas fibroblasts are decreased in number. Elastic fibers are focally crowded. There may also be a reduction in the size of the dermal appendages. 986
There is disorganization of the collagen bundles and a variable thickness of the fibers. 984 Globular masses of microfibrils are also seen.
Linear atrophoderma of Moulin was first described in 1992 and is characterized by a hyperpigmented atrophoderma that follows Blaschko’s lines.987. and 988. Telangiectatic macules were also present in one case. 989 Leukonychia has also been described. 990 Onset is usually during childhood or adolescence. The absence of sclerosis and a lilac color are distinguishing features from linear scleroderma. The lesions are similar to atrophoderma of Pasini and Pierini but lesions in this latter entity do not follow Blaschko’s lines. It has been suggested that a postzygotic mutation in lamin A is a theoretical possibility as a candidate gene.991. and 992.
The term congenital linear atrophoderma has been used for an infant with depressed, hypopigmented, linear plaques of congenital onset on the lower extremity. 993 There were some similarities to a hypopigmented and congenital form of linear atrophoderma of Moulin.
Acrodermatitis chronica atrophicans is a spirochete-induced disease (see p. 579) characterized by atrophy of the dermis to about half its normal thickness or less. The pilosebaceous follicles and subcutis also undergo atrophy.
Restrictive dermopathy (OMIM 275210) is a recently delineated, lethal, autosomal recessive disorder characterized by taut and shiny skin with facial dysmorphism, arthrogryposis multiplex, and bone dysplasia.998.999.1000.1001.1002.1003.1004. and 1005. It has also been called the tight skin contracture syndrome. Transposition of the great arteries and microcolon are two further associations. 1006
Fibroblasts show poor growth in culture, suggesting that restrictive dermopathy results from a primary abnormality in fibroblast growth and function. 998
A study in 2005 showed that restrictive dermopathy is caused by mutations either in the LMNA gene, at locus 1q21.2 encoding A-type lamins, or in the zinc metalloproteinase STE (ZMPSTE24) gene, encoding a metalloprotease essentially for the posttranslational processing of prelamin A to mature lamin A. The former defect is inherited as an autosomal dominant trait, and the latter as autosomal recessive. 1007
There is thinning of the dermis with the collagen bundles arranged parallel to the skin surface. 998 Elastic tissue may be normal or reduced in amount. Rudimentary hair follicles are usually present. There is also a reduction in adnexal structures and factor XIIIa-positive dendrocytes. The overlying epidermis is often hyperplastic with hyperkeratosis. 1003
The perforating collagenoses are a group of dermatoses in which altered collagen is eliminated from the dermis through the epidermis. 1008 Included in this group are reactive perforating collagenosis, the closely related entity of perforating verruciform ‘collagenoma’ (collagènome perforant verruciforme), and also chondrodermatitis nodularis helicis.
Collagen elimination is also found as a secondary event in some cases of granuloma annulare (perforating granuloma annulare) and of necrobiosis lipoidica. It has also been seen in healing wounds, in resolving keratoacanthomas, and following the intradermal injection of corticosteroid.1009. and 1010. Some cases do not fit neatly into any of the categories listed above. 1011
Reactive perforating collagenosis, first described in 1967, is a rare condition in which collagen is eliminated from the dermis. 1012 There are two distinct clinical variants. 1013 In the usual form, in which the onset is in childhood, there are recurrent, umbilicated papules up to 6 mm or so in diameter which spontaneously disappear after 6–8weeks, leaving a hypopigmented, sometimes scarred area. 1014 New lesions develop as older lesions are involuting and this may continue into adult life. 1015 The papules are found on the extremities, particularly the dorsum of the hands and forearms. The sole of the foot is a rare site. 1016 There is often a history of superficial trauma, such as a scratch or insect bite.1017. and 1018. Lesions have been induced experimentally by trauma.1013. and 1019. An association with Down syndrome has been reported. 1020 An autosomal recessive inheritance has been proposed for some cases.1008. and 1021.
The other clinical variant (acquired perforating collagenosis) is acquired in adult life and occurs especially in diabetics with chronic renal failure.1022.1023.1024.1025.1026.1027.1028.1029.1030. and 1031. In patients with chronic renal failure there is sometimes clinical and histological overlap with other perforating disorders such as perforating folliculitis and Kyrle’s disease.1032.1033.1034.1035.1036. and 1037. The finding that both collagen and elastic fibers undergo transepithelial elimination in these circumstances suggests that a single disease process is present. 1038 The term ‘acquired perforating dermatosis’ has been suggested for the different expressions of transepithelial elimination associated with renal disease and/or diabetes mellitus.1036.1038.1039. and 1040. ‘Giant’ lesions up to 2 cm in diameter and forming plaques in some areas have been reported. 1030 Reactive perforating collagenosis has also been reported in association with myelodysplastic syndrome, 1041 Hodgkin’s disease,1042. and 1043. internal cancers,1044. and 1045. herpes zoster,1046.1047. and 1048. the Treacher Collins syndrome, 1049 the red dye in a tattoo, 1050 lichen amyloidosus, chemical burns from commercially available salt water, 1051 Wegener’s granulomatosis, hepatitis, 1036 hypothyroidism, 1036 rheumatoid arthritis, Henoch–Schönlein purpura, 1008 and the acquired immunodeficiency syndrome associated with end-stage renal disease. 1052 Its association with scabies and atopic dermatitis may be the result of intense scratching.1053.1054.1055. and 1056.
It has been suggested that the acquired variant may be precipitated by the deposition of crystal-like microdeposits in the upper dermis, close to the site of the transepidermal channel. 1040 Such material is possibly a byproduct of the chronic renal failure.
An attempt has been made to elucidate a common mechanism for the various perforating disorders. It has been suggested that elevated serum and tissue concentrations of fibronectin (a component of the extracellular matrix) may incite epithelial proliferation and migration, culminating in perforation.1057. and 1058. TGF-β has also been implicated in this process. 1059
Cases of acquired perforating collagenosis have been successfully treated with doxycycline,1060. and 1061. allopurinol, 1030 oral acitretin, 1031 and with narrowband ultraviolet B phototherapy. 1062 Topical retinoic acid has been used to treat familial cases. 1063
The appearances vary with the stage of evolution of the lesion. In early lesions, there is acanthosis of the epidermis and an accumulation of basophilic collagen in the dermal papillae. In established lesions, there is a cup-shaped depression of the epidermis which is filled with a plug consisting of parakeratotic keratin, some collagen and inflammatory debris. The underlying epidermis is thin with fine slits through which basophilic collagen fibers in vertical orientation are extruded. Sometimes there is a complete break in the epidermis (Fig. 11.15). 1064 A few studies have shown elastic fibers in the extruded material and granulation tissue in the superficial dermis. 1065 Follicular involvement was present in 40% of cases in one series. 1063 Immunohistochemical and ultrastructural studies have shown that the extruded collagen is normal.1040. and 1065. One study has shown that the extruded collagen is type IV, suggesting origin from basement membrane. 1066 Nuclear material derived from neutrophils, and neutrophils themselves, are present in the extruded material.1067. and 1068. There is also absence of the basal lamina at the site of the perforation. 1033
Acquired perforating collagenosis in a patient with chronic renal failure. (A) There is a cup-shaped depression containing keratin, necrotic tissue, and inflammatory debris. (B) Collagen fibers are being extruded into the plug. (H & E)
Perforating verruciform ‘collagenoma’, also known as collagènome perforant verruciforme, is closely related to reactive perforating collagenosis. Traumatically altered collagen is eliminated in a single, self-limited episode.1010.1069. and 1070. The episode of trauma is usually more substantial than in reactive perforating collagenosis and the lesion that results is more verrucous. The author has seen a large plaque develop on the chest, following trauma from an automobile seat belt, with the histological features outlined below. 1071
There is prominent epithelial hyperplasia with some acanthotic downgrowths encompassing necrobiotic collagen and debris (Fig. 11.16). Elastic fibers are partly preserved in the central plug. 1034
A variant of perforating collagenosis following trauma from a seat belt in an automobile. Damaged collagen is being eliminated through the epidermal downgrowths. (H & E)
Chondrodermatitis nodularis helicis is a chronic, intermittently crusted, painful or tender nodule found primarily on the upper part of the helix of the ear of males over 50 years of age. Cases under the age of 20 years are rare.1072. and 1073. The lesions are usually solitary and average 4–6 mm in diameter. There is a predilection for the right ear in some series. 1074 The condition is infrequent in females, in whom it is more common on the antihelix. 1042 Bilateral lesions on the free border of the helix have been reported in a woman. 1075 The recurrence rate after treatment, which is usually curettage and cautery, can be as high as 20%.1076. and 1077. Lesions are now being seen as a result of chronic pressure from cell (mobile) phones. 1078 The location of the lesions correlates with the point of pressure of the phone.
The etiology and pathogenesis are speculative, but it has been suggested that the primary event is localized degeneration of dermal collagen with its subsequent partial extrusion through a central ulcer or by actual transepidermal elimination. The collagen degeneration possibly results from a combination of factors which include minor trauma or pressure (during sleep),1079. and 1080. poor vascularity, and sometimes solar damage. 1076 An autoantibody to type II collagen was present in one case. 1081 An association with the limited form of systemic sclerosis and with dermatomyositis is a rare occurrence.1082. and 1083. Magro and colleagues have reported a series of 24 patients with an underlying systemic disease including collagen vascular diseases, autoimmune thyroid disease, and non-immune-based vascular injury syndromes. 1084 The patients were characteristically younger than the usual patients with this condition. 1084 Goette has called chondrodermatitis nodularis helicis an ‘actinically induced perforating necrobiotic granuloma’, but this statement appears to be an overgeneralization. 1085 It has also been suggested that the infundibular portion of the hair follicle is primarily involved, with perforation of follicular contents into the dermis.1086. and 1087.
As already mentioned, the usual treatment is curettage and cautery, but surgical excision is often used. Punch excision of the lesion followed by a full-thickness skin graft gives results comparable with other methods. 1088 Conservative methods, aimed at removing the putative precipitating factors, namely pressure and trauma, have also been successful. 1089
The characteristic changes are a central area of ulceration or erosion or a more or less tight funnel-shaped defect in the epidermis overlying an area of dermal collagen which shows variable edema and fibrinoid degeneration (Fig. 11.17). Some inflammatory, keratinous and collagen debris caps the area of degenerate collagen, forming the crust noted clinically.
Chondrodermatitis nodularis helicis. Fibrinoid material is present in the base of an ulcer which overlies the cartilage of the ear. (H & E)
At the margins of the central defect there is variable epidermal acanthosis, which rarely assumes the proportions of pseudoepitheliomatous hyperplasia. 1087 It extends peripherally over 3–5 rete pegs and there is usually a prominent granular layer with some overlying hyperkeratotic and parakeratotic scale. 1091 Richly vascularized granulation tissue borders the necrobiotic material peripherally and sometimes the vessels have some glomus-like features. This area usually contains a mild, but sometimes moderately heavy inflammatory cell infiltrate. The infiltrate is predominantly lymphocytic with an admixture of plasma cells, histiocytes and sometimes a few neutrophils. Rarely, the histiocytes may assume a palisaded arrangement at the margins of the necrobiotic zone. 1091 Irregular slit-like spaces may extend into the degenerate collagen, and other spaces containing fibrin may be found at the dermoepidermal junction above the peripheral granulation tissue. Uncommonly, degenerate collagen will be seen in slits within the epidermis, representing true transepidermal elimination.
Beyond the lesion itself there may be some telangiectatic vessels in the upper dermis and variable solar elastosis. 1085 Elastic fibers are diminished and focally absent in the area of degenerate collagen. Nerve hyperplasia is present in most lesions, the possible mechanism of the pain that occurs with light pressure. 1092 This change is often masked by the intense vascular and inflammatory reactions. 1092
In nearly all cases there are changes in the perichondrium which are most marked directly beneath the degenerate collagen. These changes include fibrous thickening and very mild chronic inflammation. Degenerative changes may also be found in the cartilage, with alterations in its staining qualities, patchy hyalinization, and, uncommonly, partial destruction with necrosis. 1091 Calcification and even ossification have been noted in the distal part of the chondral lamina. 1093 In healed lesions there is dermal fibrosis which also involves the perichondrium (Fig. 11.18).
Chondrodermatitis nodularis helicis. This healed lesion shows fibrosis of the dermis and perichondrium. (H & E)
Variable changes in dermal collagen are found in Ehlers–Danlos syndrome, osteogenesis imperfecta, and Marfan’s syndrome. Marked thinning of the dermis can occur in some forms of all three disorders, although in other clinical variants it may appear quite normal. Defects in the biosynthesis of collagen have been detected in a number of patients with osteogenesis imperfecta and Ehlers–Danlos syndrome, although in the majority of cases the defect has not been defined. 1
Ehlers–Danlos syndrome is a heterogeneous disorder of connective tissue which combines hyperextensible fragile skin and loose-jointedness with a tendency to bruising and bleeding.1094.1095.1096.1097.1098. and 1099. Absence of the inferior labial and lingual frenula is associated with the classical and hypermobile types. 1100 At least 11 types are now recognized, each with a characteristic clinical expression, inheritance pattern, and, in some cases, a defined defect.9. and 1101. Paradoxically, specific defects in collagen biosynthesis have not been elucidated in the common forms of this disease. 1102 Syndrome delineation is still proceeding and many cases do not fit neatly into any category. 1103 In 1997, a revision of the traditional classification used below was proposed, based primarily on the cause of each type. 1104 This classification, which has not gained wide acceptance, is shown in Table 11.2.
Gravis (type I)
Mitis (type II)
X-linked (type V)
Periodontitis (type VIII)
Fibronectin-deficient (type X)
Familial hypermobility (type XI)
Progeroid
Contradictory results have been published on the histopathology of this condition.1105. and 1106. More recent reports have generally stated that the dermis is normal on light microscopy. 1105 Marked thinning of the dermis has been reported in some variants of type IV16 and also in type VI, while solar elastotic changes have been documented in type IV disease. 1105 Elastosis perforans serpiginosa has been reported in several cases of the syndrome, again particularly type IV. 1008 Similarly, ultrastructural studies have been contradictory, with some authors reporting normal fiber diameter and others observing some variability in size.16.1107. and 1108. A distorted arrangement of fibrils has been noted in several reports. 1107 Scanning electron microscopy may offer further information in the future, as preliminary studies have shown disordered fibril aggregation and orientation. 1096 Other ultrastructural findings will be mentioned below in the discussion of the specific types of the syndrome.
Patients with types I (OMIM 130000) and II (OMIM 130010) Ehlers–Danlos syndrome are now referred to as having the classic type. Clinical features are usually severe in this common, autosomal dominant (gravis) form of the disease. 4 Herniation of fat into the dermis, with subsequent calcification, may occur at pressure points. 1109 Increased fiber diameter and loosely assembled fibrils have been reported. 1110 The biochemical defect is not known, although reduced type III collagen production has been reported in one case. 1110 Mutations have been found in the COL5A1, COL5A2, and tenascin-X genes in a limited number of patients/families.12.1111. and 1112. Failure to detect a defect in many patients with types I/II may be due to so-called ‘haploinsufficiency’ (loss of expression of one allele) which is present in up to one-third of individuals with this classic type. 1113 An arginine → cysteine substitution in type I collagen has also been reported in a family. This finding, which implicates another fibrillar collagen, confirms the genetic heterogeneity of this type. 1114
There is a reduction in the number of factor XIIIa-positive dendrocytes in the classic type (types I and II of the traditional classification system). There is also a reduction in skin thickness on ultrasound examination. 1115
The molecular defect in this mild, autosomal dominant form is unknown. 4 Partial deletion of the C-terminal end of the procollagen molecule has been demonstrated in one case. 6 Minor variations in fibril diameter have been noted ultrastructurally, and in one case there was lateral fusion of fibrils. 1108 It has been suggested that mild variants of the mitis form are not uncommon in the general population.1116. and 1117.
In type III (OMIM 130020), there is marked joint hypermobility but only minor skin changes.4. and 1118. Skin thickness is reduced on ultrasound examination. 1115 Inheritance is autosomal dominant. Some cases are due to a deficit of tenascin-X, but no molecular alteration has been described in the majority of cases. Elastic fibers in the tenascin-X cases are fragmented and clumped. 1119 A recessive variant of this syndrome has been related to a deficiency of tenascin-X. 1120
Type IV is a heterogeneous group with at least four subtypes.1121. and 1122. Both autosomal dominant (OMIM 130050) and recessive variants occur.1121. and 1123. There is a reduced life expectancy because of a propensity for rupture of large arteries and internal hollow viscera, particularly the colon.9. and 1124. Thin, translucent skin on the face and distal parts of the limbs in several subtypes gives the appearance of premature aging (acrogeria –p. 329).1122.1125. and 1126. Type IV results from a mutation in the COL3A1 gene that controls type III collagen synthesis.1102.1127. and 1128. It is located in the region of chromosome 2q31. 1129 A decreased amount of type III procollagen is recovered from cultured skin fibroblasts.9.1130. and 1131. Defective type III collagen is sometimes found. 9 However, a case with type III collagen deficiency but with a normal phenotype has been documented. 1132
Vascular rupture has also been reported in patients with type I collagen R-to-C substitution, indicating the difficulty of classifying some cases with the traditional classification system. 1133
Ultrastructural changes have included the finding of collagen fibrils with reduced diameter, the presence of dilated rough endoplasmic reticulum in fibroblasts, and fragmentation of elastic fibers. 1125
Immunofluorescence of cultured skin fibroblasts has shown abnormal amounts of type III collagen retained in their cytoplasm. 1134 This is not a feature in normal subjects or in other connective tissue diseases.
The presence of thin skin can be confirmed by ultrasound. 1135
This autosomal recessive variant (OMIM 225400) is characterized by ocular fragility, kyphoscoliosis, and prominent cutaneous and joint signs.4. and 1137. There is a deficiency in peptidyl lysyl hydroxylase and an absence of hydroxylysine in dermal collagen.1094. and 1138. This results in abnormal crosslinks in both type I and type III collagen. 1139 The candidate gene is PLOD1, which maps to chromosome 1p36.3–p36.2. This variant of the syndrome is probably a biochemically heterogeneous entity, as normal levels of enzyme have been found in some individuals. 4
There is gross joint instability leading to multiple dislocations at birth. 1140 There are three subtypes, all of which are characterized by defects in the conversion of type I procollagen to collagen. 1102 In types A and B, there are abnormalities in the N-terminal procollagen peptidase cleavage sites.1131. and 1141. A deletion in chromosome 6q has been reported in one patient with type VIIB (OMIM 130060). 1142 A third subgroup, type VIIC, results from deficient procollagen I N-proteinase. 1143 It results from inactivating mutations in the ADAMTS-2 gene. 1144 It is similar to dermatosparaxis in cattle.1143.1145.1146. and 1147. On electron microscopy there are small, irregular, and circular collagen fibers in the skin. 1143
The type VIII autosomal dominant form (OMIM 130080) is marked by easy bruising, pretibial hyperpigmented scars, and the early onset of periodontal disease.1094.1148. and 1149. Periodontal disease is unique to this variant. 1150 There may be loose-jointedness of the fingers. The molecular defect is currently unknown but the gene appears to be located at 12p13.1150. and 1151. The microfibril-associated glycoprotein-2 gene (MAGP2) is a promising candidate gene. 1149 A reduction in collagen type I and III synthesis was detected in a recent case. 1152 The dermis often appears normal on light microscopy.
The type IX variant is X-linked and characterized by the presence of occipital horns and other skeletal abnormalities and a propensity to bladder diverticula and to hernias.1136. and 1153. Cutaneous changes are mild. There is defective activity of lysyl oxidase. It has been suggested that all disorders with deficient lysyl oxidase, including Menkes’ syndrome (see p. 349), should be classified as type IX disease,1154. and 1155. but this ignores the absence of the usual clinical features of Ehlers–Danlos syndrome in patients with Menkes’ syndrome.
Some cases of the type X variant (OMIM 225310) were originally classified as type IX disease. 1156 There are moderately severe skin and joint changes and a defect in platelet aggregation. 1156 The platelet defect can be corrected by fibronectin, which is an important adhesive glycoprotein that crosslinks to collagen. 1156
Many cases of Ehlers–Danlos syndrome defy classification, such as the case reported with abnormalities localized to the shoulder region1157 and another with intracellular degradation of alpha-2 chains of type I procollagen. 1158 This same defect is found in some cases of osteogenesis imperfecta and of Marfan’s syndrome. 1158 Mutations in genes encoding types I and II transforming growth factor-β (TGFBR1) and (TGFBR2) cause the newly recognized Loeys–Dietz syndrome, which is characterized by aortic aneurysms, generalized arterial tortuosity, and a variety of craniofacial and skeletal anomalies. 1149
It has recently been suggested that women with recurrent preterm premature rupture of fetal membranes may have a form of Ehlers–Danlos syndrome as examination of their skin shows a thinned dermis, with thinned collagen bundles and some ribbon-like elastic fibers. 1159
Bone fragility is the cardinal manifestation of osteogenesis imperfecta (OMIM 166200), a genetically heterogeneous entity in which various defects in type I collagen have been detected.7. and 1160. Other clinical manifestations include short stature, joint laxity, blue sclerae, and otosclerosis. The skin may be thin in the severe variants; it has reduced elasticity. 1161
The dermis is markedly reduced in thickness in those with clinically thin skin. 16 The fine collagen bundles which constitute the dermis are often argyrophilic.
The clinical features of Marfan’s syndrome (OMIM 154700) include tall stature, skeletal malformations, arachnodactyly, and dislocation of the lens.1162. and 1163. It is caused by mutations in the fibrillin-1 gene (FBN1). 1164 Cutaneous manifestations are relatively insignificant. Marfan’s syndrome is discussed further in Chapter 12, page 350.
In some cases, thinning of the dermis, resulting from a diminished quantity of collagen and thinner collagen bundles, has been reported. This has been confirmed ultrastructurally. An increase in matrix material and fragmentation of elastic fibers have also been reported.
The syndromes of premature aging can be a very small or quite large disease group, depending on the criteria used.1165. and 1166. There are five conditions with prominent cutaneous findings that are usually included in any discussion on this subject – Werner’s syndrome (adult progeria), progeria (Hutchinson–Gilford syndrome), acrogeria, Rothmund–Thomson syndrome (poikiloderma congenitale), and Cockayne’s syndrome.1165. and 1167. The Rothmund–Thomson syndrome includes poikiloderma as a major feature and is accordingly discussed with the lichenoid reaction pattern (see p. 67), while in Cockayne’s syndrome photosensitivity and sensitivity of cultured fibroblasts to ultraviolet light is a major feature. The other three syndromes show changes in dermal collagen and accordingly they will be considered further in this chapter. In the two types of progeria this takes the form of sclerodermoid lesions, while in acrogeria the dermis is almost always atrophic.
In addition to the five syndromes listed, premature aging is also a feature of Down syndrome. 1168 The capacity to repair UV-induced DNA damage is diminished in this syndrome and this deteriorates further with aging. 1168 The Wiedemann–Rautenstrauch syndrome (OMIM 264090) is a further exceedingly rare progeroid syndrome present at birth. 1169
Werner’s syndrome (OMIM 277700) is a rare, autosomal recessive disorder in which evidence of premature aging becomes manifest between the ages of 15 and 30 years.1165. and 1170. It is common in Japan. 1171The clinical features include short stature, bird-like facies, juvenile cataracts, a tendency to diabetes mellitus, trophic ulcers of the legs, premature graying of the hair and balding, an increased incidence of neoplasms, and acral sclerodermoid changes.1165.1172.1173.1174. and 1175. Diffuse lentiginosis has been recorded in one case. 1176 Extensive tendon calcification and sacroiliitis have been reported in a brother and sister with this syndrome. 1177 Death usually occurs in the fifth decade from the complications of arteriosclerosis.
Skin fibroblasts from patients with Werner’s syndrome are difficult to culture, with a slow growth rate and short life span,1178. and 1179. although in one study they produced more collagen but less glycosaminoglycans than normal. 1180 Studies have confirmed that cultured fibroblasts produce increased amounts of some collagen, but not type VI; 1181 the levels of type I and III collagen mRNA are increased, suggesting an alteration in the control of collagen synthesis at the transcriptional level. 1182 Fibroblasts appear to become hyporesponsive in vitro to a stimulator of collagen synthesis that has been found in the serum of patients with Werner’s syndrome. 1183 The pathogenetic significance of these disparate findings remains to be elucidated. Cells in Werner’s syndrome may have subtle defects in DNA repair. 1184
It results from mutational inactivation of the human RECQL2 (helicase) gene found at 8p11–p12.1179.1185.1186. and 1187. It is also known as the WRN gene. 1188 Other conditions resulting from mutations of helicase genes include Bloom’s syndrome (see p. 67) and Rothmund–Thomson syndrome (see p. 67). 1185 Chromosomal analyses on cultured fibroblasts reveal that aberrations occur frequently and randomly. 1189
There is usually some epidermal atrophy, although there may be hyperkeratosis over bony prominences. 1172 In the scleroderma-like areas there is variable hyalinization of the thickened dermal collagen. There may be replacement of subcutaneous fat by connective tissue. The pilosebaceous structures and sweat glands become atrophic. Calcinosis cutis may also develop. 1183
The Hutchinson–Gilford progeria syndrome (OMIM 176670) is a rare disease with markedly accelerated aging, clinical features becoming apparent in the first year of life. 1166 Its estimated incidence is 1 per 4–8 million live births. 1191 These features include growth retardation, alopecia, craniofacial disproportion, loss of subcutaneous fat, and atrophy of muscle.1165.1192.1193. and 1194. The skin is generally thin as a result of the loss of subcutaneous fat, except for areas with scleroderma-like plaques. 1195 Acral hyperplastic scars and keloid-like nodules composed of type IV collagen have also been described. 1196 Extensive cutaneous calcinosis may develop. To date, mutations in two genes, LMNA and ZMPSTE24, have been found in this syndrome. 1191 Most reports have implicated the lamin A/C (LMNA) gene essential for the synthesis of lamin A from prelamin A. Lamin A is involved in DNA replication, RNA transcription, and other chromatin functions. It is also involved in cell growth and cell death. 1197 The same genes are involved in restrictive dermopathy (see p. 324). 1166 A review of the laminopathies resulting from mutations in the LMNA gene was published in 2006. 1198
The biochemical basis of this disease is unknown, but it has been shown that skin fibroblasts are difficult to culture and tropoelastin production by fibroblasts is markedly increased. 1192
The scleroderma-like plaques show a diffusely thickened dermis with hyalinization of collagen. 1199 This may assume a homogenized appearance in the lower dermis. Fibrous tissue often extends into the subcutis. Hair follicles are atrophic and eventually lost. 1200 There are variable changes in the elastic tissue. In other areas of the body there is loss of subcutaneous fat and some atrophy of the dermis. 1199
Acrogeria (Gottron’s syndrome) is an exceedingly rare disease (OMIM 201200), with onset in early childhood. 1201 There is atrophy, dryness, and wrinkling of the skin which is most severe on the face and extremities.1165.1167. and 1201. Interesting associations include bony abnormalities and disorders of dermal elastic tissue in the form of elastosis perforans serpiginosa and perforating elastomas.1201.1202. and 1203. Vascular lesions such as occlusion of the digital arteries and perniosis have been reported in one patient. 1204 Inheritance is probably autosomal recessive. A mutation in the COL3A1 gene, located at chromosome 2q31–q32, has been reported in several cases.1129. and 1205. A syndrome described as metageria has features that overlap with those of acrogeria.1165. and 1167. Acrogeria is regarded by some as a distinct subgroup of type IV Ehlers–Danlos syndrome. 1206
There is atrophy of the dermis with degenerative changes in the collagen. Fibers may be swollen and present a ‘boiled’ appearance. The subcutaneous fat is often replaced by connective tissue which is indistinguishable from the dermal collagen. Elastic fibers are disrupted and irregular, with some clumping. Changes of elastosis perforans serpiginosa may be present.
Introduction
1. Spranger, J, The developmental pathology of collagens in humans, Birth Defects 23 (1987) 1–16.
2. Uitto, J; Lichtenstein, JR, Defects in the biochemistry of collagen in diseases of connective tissue, J Invest Dermatol 66 (1976) 59–79.
3. Burgeson, RE, Genetic heterogeneity of collagen, J Invest Dermatol 79 (1982) 25s–30s.
4. Krieg, T; Ihme, A; Weber, L; et al., Molecular defects of collagen metabolism in the Ehlers Danlos syndrome, Int J Dermatol 20 (1981) 415–425.
5. Prockop, DJ; Kivirikko, KI, Heritable diseases of collagen, N Engl J Med 311 (1984) 376–386.
6. Pope, FM; Dorling, J; Nicholls, AC; Webb, J, Molecular abnormalities of collagen: a review, J R Soc Med 76 (1983) 1050–1062.
7. Tsipouras, P; Ramirez, F, Genetic disorders of collagen, J Med Genet 24 (1987) 2–8.
8. O’Toole, EA, Extracellular matrix and keratinocyte migration, Clin Exp Dermatol 26 (2001) 525–530.
9. Stolle, CA; Pyeritz, RE; Myers, JC; Prockop, DJ, Synthesis of an altered type III procollagen in a patient with type IV Ehlers–Danlos syndrome, J Biol Chem 260 (1985) 1937–1944.
10. Stanley, JR; Woodley, DT; Katz, SI; Martin, GR, Structure and function of basement membrane, J Invest Dermatol 79 (1982) 69s–72s.
11. Sage, H, Collagens of basement membrane, J Invest Dermatol 79 (1982) 51s–59s.
12. Giunta, C; Steinmann, B, Compound heterozygosity for a disease-causing G1489D and disease-modifying G530S substitution in COL5A1 of a patient with the classical type of Ehlers–Danlos syndrome: an explanation of intrafamilial variability? Am J Med Genet 90 (2000) 72–79.
13. Akagi, A; Tajima, S; Ishibashi, A; et al., Expression of type XVI collagen in human skin fibroblasts: enhanced expression in fibrotic skin diseases, J Invest Dermatol 113 (1999) 246–250.
14. Uitto, J; Tan, EML; Ryhanen, L, Inhibition of collagen accumulation in fibrotic processes: review of pharmacologic agents and new approaches with amino acids and their analogues, J Invest Dermatol 79 (1982) 113s–120s.
15. Prockop, DJ; Kivirikko, KI; Tuderman, L; Guzman, NA, The biosynthesis of collagen and its disorders, N Engl J Med 301 (1979) 13–23.
16. Holbrook, KA; Byers, PH, Structural abnormalities in the dermal collagen and elastic matrix from the skin of patients with inherited connective tissue disorders, J Invest Dermatol 79 (1982) 7s–16s.
Scleroderma
17. Young Jr, EM; Barr, RJ, Sclerosing dermatoses, J Cutan Pathol 12 (1985) 426–441.
18. Rodnan, GP; Jablonska, S; Medsger Jr, TA, Classification and nomenclature of progressive systemic sclerosis, Clin Rheum Dis 5 (1979) 5–12.
19. Callen, JP; Provost, TT; Tuffanelli, DL, Periodic synopsis on collagen vascular disease, J Am Acad Dermatol 13 (1985) 809–815.
20. LeRoy, EC; Black, C; Fleischmajer, R; et al., Scleroderma (systemic sclerosis): classification, subsets and pathogenesis, J Rheumatol 15 (1988) 202–205.
21. Krafchik, BR, Localized cutaneous scleroderma, Semin Dermatol 11 (1992) 65–72.
22. Jablonska, S; Rodnan, GP, Localized forms of scleroderma, Clin Rheum Dis 5 (1979) 215–241.
23. Kyriakis, KP; Emmanuelides, S; Terzoudi, S; et al., Gender and age prevalence distributions of morphea en plaque and anogenital lichen sclerosus, J Eur Acad Dermatol Venereol 21 (2007) 825–826.
24. Barba, A; Rosina, P; Chieregato, C; D’Onghia, FS, Morphoea in a newborn boy, Br J Dermatol 140 (1999) 365–366.
25. Maricq, HR, Capillary abnormalities, Raynaud’s phenomenon, and systemic sclerosis in patients with localized scleroderma, Arch Dermatol 128 (1992) 630–632.
26. Ruffatti, A; Peserico, A; Glorioso, S; et al., Anticentromere antibody in localized scleroderma, J Am Acad Dermatol 15 (1986) 637–642.
27. Weinberg, JM; Russo, M; Hirsch, RJ; Don, PC, Morphoea of the breast in a young girl, Clin Exp Dermatol 26 (2001) 497–498.
28. Serup, J, Clinical appearance of skin lesions and disturbances of pigmentation in localized scleroderma, Acta Derm Venereol 64 (1984) 485–492.
29. Synkowski, DR; Lobitz Jr, WC; Provost, TT, Bullous scleroderma, Arch Dermatol 117 (1981) 135–137.
30. Peterson, LS; Nelson, AM; Su, WPD, Classification of morphea (localized scleroderma), Mayo Clin Proc 70 (1995) 1068–1076.
31. Sehgal, VN; Srivastava, G; Aggarwal, AK; et al., Localized scleroderma/morphea, Int J Dermatol 41 (2002) 467–475.
32. Winkelmann, RK, Localized cutaneous scleroderma, Semin Dermatol 4 (1985) 90–103.
33. Leibovici, V; Zlotogorski, A; Kanner, A; Shinar, E, Generalized morphea and idiopathic thrombocytopenia, J Am Acad Dermatol 18 (1988) 1194–1196.
34. Cantwell Jr, AR; Jones, JE; Kelso, DW, Pleomorphic, variably acid-fast bacteria in an adult patient with disabling pansclerotic morphea, Arch Dermatol 120 (1983) 656–661.
35. Diaz-Perez, JL; Connolly, SM; Winkelmann, RK, Disabling pansclerotic morphea of children, Arch Dermatol 116 (1980) 169–173.
36. Maragh, SH; Davis, MDP; Bruce, AJ; Nelson, AM, Disabling pansclerotic morphea: Clinical presentation in two adults, J Am Acad Dermatol 53 (2005) S115–119.
37. Herzinger, T; Prinz, JC; Röcken, M, Pansclerotic morphea of the head, Arch Dermatol 144 (2008) 125–126.
38. Nguyen, XH; Hansen, R; Valencia, F, Severe ankle deformity secondary to pansclerotic morphea in a 9-year-old girl: correction involving arthrodesis and free flap coverage, Pediatr Dermatol 19 (2002) 560–563.
39. Parodi, PC; Riberti, C; Draganic Stinco, D; et al., Squamous cell carcinoma arising in a patient with long-standing pansclerotic morphea, Br J Dermatol 144 (2001) 417–419.
40. Wollina, U; Buslau, M; Weyers, W, Squamous cell carcinoma in pansclerotic morphea of childhood, Pediatr Dermatol 19 (2002) 151–154.
41. Doyle, JA; Connolly, SM; Winkelmann, RK, Cutaneous and subcutaneous inflammatory sclerosis syndromes, Arch Dermatol 118 (1982) 886–890.
42. Whittaker, SJ; Smith, NP; Russell Jones, R, Solitary morphoea profunda, Br J Dermatol 120 (1989) 431–440.