Fig. 8.1
Shiny, hypopigmented plaques with satellite plaques over the gluteal fold, representing superficial dermal chronic graft-versus-host disease (cGVHD) of the lichen sclerosus type
Deep Dermis: Morpheaform Type
The classic sclerotic-type (morpheaform) cGVHD tends to occur deeper in the dermis and thus does not result in the superficial hypopigmentation or cigarette-paper atrophy (Fig. 8.2). However, both superficial and deep forms of these entities may coexist. It tends to occur de novo in areas of pressure and chronic friction, such as the waistband (Fig. 8.3), the axilla, and areas of chronic edema, such as the lower extremity. Other than areas of edema and chronic pressure, there are also many reports of the isotopic and isomorphic development within areas of trauma or preexisting cutaneous damage, such as herpess zoster (Fig. 8.4), port sites, or previous needle-sticks [6, 7]. The similarities to morphea are very interesting, as there are also many reports of morphea developing within areas of localized cutaneous radiotherapy [8]. A specific entity that tends to develop out of morpheaform cGVHD is the angiomatosis phenomenon (Fig. 8.5) [9]. These patients present with long-standing sclerosis with superimposed vascular patches or even nodules that may look like dense telangiectasias to very large pyogenic granulomas.
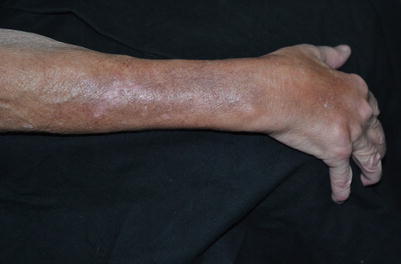
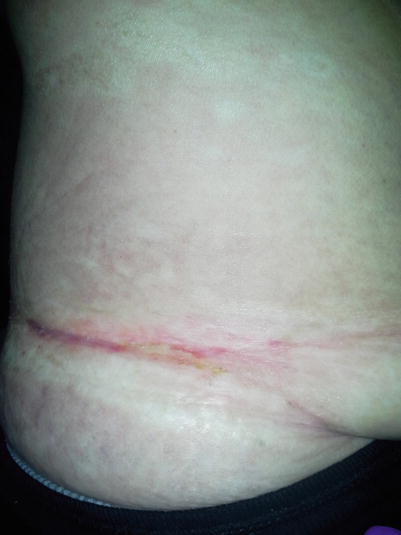
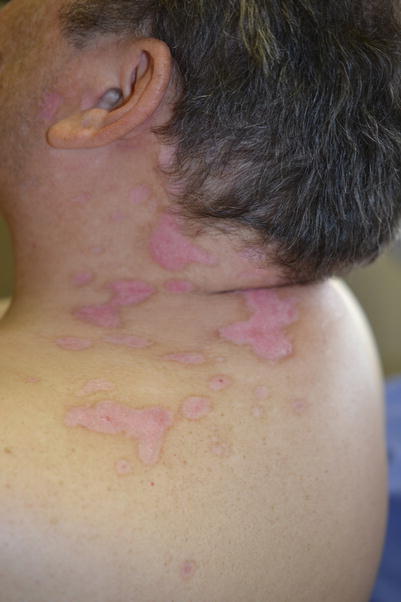
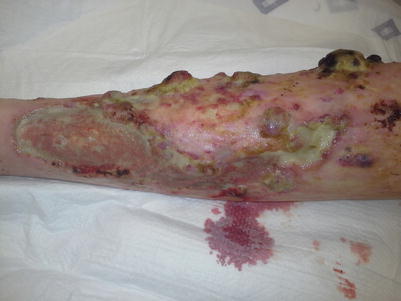
Fig. 8.2
The right forearm of a patient with extensive induration over the dorsal arms, representing deeper dermal involvement; superficial hypopigmentation is no longer visible
Fig. 8.3
Sclerotic-type cGVHD developing at the site of the waistband, representing chronic friction, with features of both morpheaform and lichen sclerosus morphologies, as well as a central ulceration
Fig. 8.4
Infiltrated hypopigmented plaques developing at the site of previous herpes zoster
Fig. 8.5
Extensive vascular plaques and tumors developing over the lower extremity, interspersed with large ulcerations at the site of previous plaques, occurring within long-standing sclerotic-type cGVHD and representing the GVHD-angiomatosis phenomenon
Subcutaneous Fat (Panniculitic) cGVHD
The next layer of the integument is the subcutaneous fat or the panniculus. The typical autoimmune or autoinflammatory diseases that affect this area are called panniculitis, with examples such as erythema nodosum, lupus panniculitis, or erythema induratum. Subcutaneous involvement of cGVHD typically has an appearance that would best fit a severe form of lupus panniculitis. The evanescent erythema and indurated patches typical for erythema nodosum are not present; instead, the skin has normal coloration with extensive underlying rippling, resembling a cobblestone appearance or accentuated cellulite. The architecture of the subcutaneous fat consists of numerous lobules that are held in place by fibrous septae that encompass the lobules and provide structure. cGVHD in this setting appears to thicken the septae, resulting in a regularly spaced fat herniations surrounded by tethered septae (Fig. 8.6).
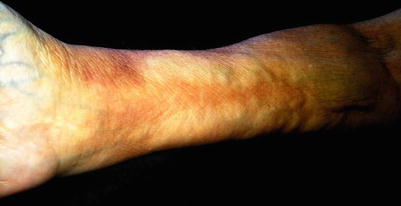
Fig. 8.6
Infiltrated plaques diffusely over the distal arm, with the proximal arm developing a cobblestoning or “pseudocellulite” appearance from the involvement of the subcutaneous fat
Fascial cGVHD
Although the fascia is of musculoskeletal derivation and is not part of the integument, there is often fibrosis adherent to this layer. The prototype autoimmune disease of this level is eosinophilic fasciitis or Shulman syndrome. The fascial disease in cGVHD does not have an age or sex predilection, and the findings may be unnoticed initially except for a subtle groove sign, indicating the tethering of the skin along the tendons (Fig. 8.7). The other signs of involvement include the restricted prayer sign (Fig. 8.8) and the pipestem deformity (Fig. 8.9). This last finding tends to indicate that the disease has progressed to involve not only fascia but full-thickness to the superficial layers of the dermis. At this point, the skin is diffusely infiltrated and immobile from the underlying muscle or bone.
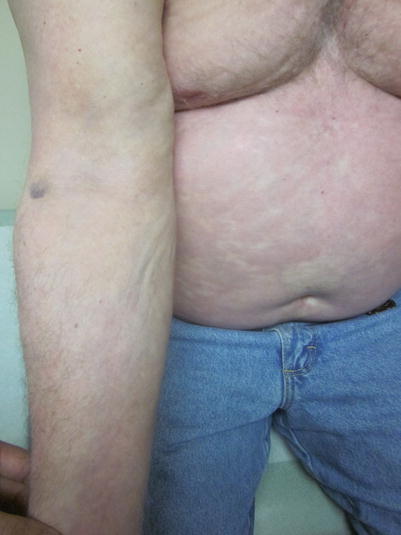
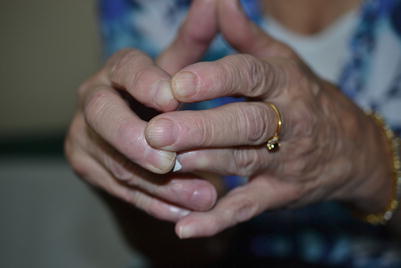
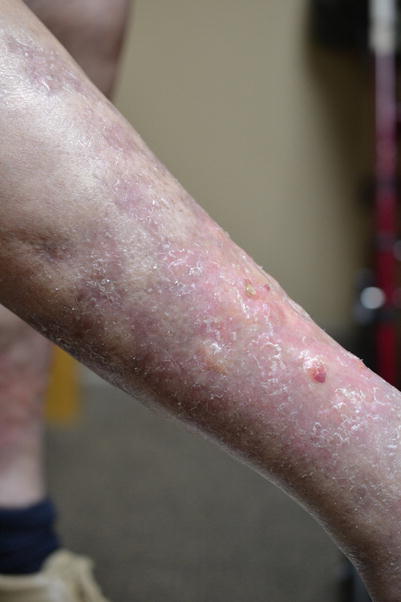
Fig. 8.7
Linear groove over the right forearm flexors, forming the “groove sign” of fascial involvement of sclerotic-type cGVHD
Fig. 8.8
The impaired “prayer sign” preventing the patient from flatly apposing her hands, indicating fascial disease of her forearms
Fig. 8.9
Diffuse involvement over lower extremity from the superficial dermis to the subcutaneous fat and fascia, resulting in the “pipestem” deformity. This patient also has an incidental findings of GVHD angiomatosis
Prognosis
When comparing patients with all forms of cGVHD to those without cGVHD, there is reduced overall and nonrelapse survival [10–12], although mild disease does not appear to impact survival. Consistently, higher mortality has been shown in patients with all types of cGVHD, when comparing severe to mild [13]. When looking at the impact of body surface area involvement in patients with sclerotic-type cGVHD, mortality is higher for those with more extensive sclerotic disease [5].
Although there is an impaired survival rate in patients with moderate or severe sclerotic-type cGVHD, another significant issue is the loss of quality of life (QOL) from associated lung disease, contractures, immobility, pruritus, ulcerations, and pain. Patients with cGVHD have been shown to have an overall QOL similar to patients with other severe autoimmune diseases such as multiple sclerosis, systemic sclerosis, and systemic lupus erythematosus [14]. Studies have shown that about 40 % of patients who develop cGVHD will be on disability or medical leave because of their illness [13]. Specifically, patients with sclerotic-type cGVHD tend to complain of more restriction of movement, loss of grip strength, skin color changes, skin ulcerations, itching, and joint stiffness than patients with other forms of GVHD [5].
Treatment
Treatments can be subdivided into those that are skin-directed and those that are systemic. The ultimate treatment decision will depend on more than just cutaneous involvement, especially if higher-risk organs such as the GI tract, lungs, and liver are also involved. Patients with mild disease may not need treatment; they may be benefiting from some graft-versus-leukemia effect at that stage [15].
Scoring Systems to Determine the Effect of Treatment
One of the difficulties in assessing the prognosis and QOL features is that cGVHD is a multiorgan disease. It is difficult to assess sclerotic-type skin disease alone without the effect of oral, genital, ocular, or lung disease. However, the discussion of treatment efficacy will attempt to focus on studies using cutaneous endpoints.
The most commonly used validation system is the National Institutes of Health (NIH) consensus conference criteria, based on a simple four-point scale to assess a patient for no involvement or mild, moderate, or severe involvement [15]. This system was initially created on the basis of expert opinion, and although it appears very simplistic, developing stronger, organ-specific staging methods has not proved better in predicting disease mortality [16]. As no scales have yet surpassed this system, it is usually used for clinical trials, often supplemented with QOL measurements. The QOL tools typically used are the Lee symptom scale, a validated rating specifically for patients with cGVHD [13], and the SF-36 Short Form Health Survey, among others.
Skin-Directed Therapeutics
Topical Corticosteroids, Vitamin D Analogues, Calcineurin Inhibitors
There are very limited data on topical immune modulators (which have indications for psoriasis and atopic dermatitis), and their expected benefit for patients with deep dermal disease is low, but they do offer the benefit of being inexpensive and generally low-risk. In a case series of 18 patients with cGVHD, 13 of the 18 found a benefit from topical tacrolimus in terms of scaling, pain, or “tightness,” but validated response scales were not used and the patients did not necessarily have the sclerotic disease [17]. For patients with superficial dermal disease (ie, lichen sclerosus–like disease) topical corticosteroids and calcineurin inhibitors may have a particular benefit. Topical corticosteroids and calcineurin inhibitors are the standard of care for patients with lichen sclerosus. A typical treatment consists of a super-high-potency corticosteroid such as augmented betamethasone dipropionate 0.05 % or clobetasol 0.05 % cream/ointment applied twice daily to the affected area, then transitioning to tacrolimus 0.1 % ointment as the patient improves. The transition to tacrolimus ointment is important, to prevent concerns such as worsening of dermal atrophy, telangiectasias, and development of striae. One should be wary of occluding the tacrolimus ointment for long periods of time or over large surface areas, as systemic absorption has occurred in patients with cGVHD. Using lichen sclerosus as a model, it appears very reasonable to treat patients with superficial cutaneous cGVHD initially with topical immunomodulators. For patients with deeper disease, one can try to use topical immunomodulator to manage the associated pruritus.
Phototherapy
Based on the action spectrum of available phototherapy wavelengths, ultraviolet A (UVA) spectrum with the longer wavelengths (320–400 nm) would appear more effective for deeper disease. To maximize the strength of the UVA and minimize the amount of required time for patients, patients may ingest 8-methoxypsoralen (psoralen + UVA = PUVA or photochemotherapy) or have the same compounded into a 1 % topical ointment to be applied 30–60 min prior to the phototherapy. The other commonly used light spectrum is narrow-band ultraviolet B (nbUVB, 311–313 nm). The advantage of this wavelength is that it is shorter, with a decreased risk of melanoma and nonmelanoma skin cancers. With the shorter wavelength, however, it is generally considered less able to treat deep skin disease. Both treatments work through inactivation of antigen-presenting cells and activated T cells, while building antigen tolerance over time. One side benefit from instituting phototherapy is the reduction in itch, although it generally takes about 2 months for improvement. A systematic review has suggested a benefit from both UVA1 and PUVA for patients being treated for sclerotic-type cGVHD as well as for the prototype diseases—lichen sclerosus, morphea, and eosinophilic fasciitis [18].
Narrow-Band UVB
Studies in cGVHD are only case series, but there is a report of two patients with sclerotic-type cGVHD who noticed a substantial improvement in pruritus and dryness of their skin after 30–40 treatments using nbUVB, without benefit to their deep sclerosis [19]. Conversely, recent reports suggest that at least one patient with sclerotic-type cGVHD has shown improvement from the use of nbUVB, but the cumulative dose energy was twice as much as the previous report [20]. At this time, there is sparse evidence to suggest a benefit for patients with sclerotic forms of cGVHD, but given the low cost, minimal adverse effects, and clear benefit in patients with acute and overlap forms of GVHD, a trial for 2–3 months may be reasonable.
Photochemotherapy/PUVA
The active medicine with this technique is 8-methoxypsoralen, the same chemical used in extracorporeal photopheresis. As mentioned previously, UVA with a longer wavelength penetrates well into the dermis. In patients who had the lichen sclerosus subtype, using PUVA in combination with multiple systemic therapies resulted in some improvement in two patients and significant improvement in three patients treated with 20–160 treatments [21]. Another three patients showed significant objective improvement by depth of fibrosis using PUVA, as did one patient with nbUVB and one with UVA1 [22]. PUVA appears effective for some patients, but even if a strong response is generated, these patients should ensure a follow-up with dermatology at least once yearly, as there is an increased risk of melanoma and nonmelanoma skin cancer.
UVA1
UVA1 is a mid-range wavelength that is administered for sclerotic diseases without concomitant ingestion or application of psoralen. It is used primarily for patients with sclerotic disease such as morphea. The greatest limitation to its use is finding dermatologists who have the device, which tends to be very expensive and requires long treatment sessions. Several single-patient case reports suggest efficacy in highly pretreated patients [22, 23]. Studies have shown complete remission in three of five patients with sclerotic-type cGVHD and partial improvement in two of five patients in an average of 21 sessions [24]. Another study showed that in patients with sclerotic-type disease, all three patients had a partial response to UVA1 treatments, with all being able to reduce their immunosuppression [25].
Non–skin-directed Systemic Therapeutics
Systemic therapeutics are used to suppress graft activity, although the ideal treatment would be one that results in modulation of graft-versus-host activity without loss of the graft-versus-leukemia/lymphoma effect.
Conventional Immunosuppression, Corticosteroids, Steroid-Sparing Immunosuppressants, Calcineurin Inhibitors
High-dose (1 mg/kg) oral corticosteroids and steroid-sparing agents such as mycophenolate mofetil make up the first-line standard of care for patients with moderate or severe cGVHD. If patients improve, then the corticosteroids are weaned. In practice, if the patient’s GVHD is skin-predominant, utilizing a skin-directed therapy such as PUVA or UVA1 or an immunomodulator such as extracorporeal photopheresis (ECP) is a reasonable option to help wean systemic corticosteroids. If steroids fail, there is no clear evidence-based escalation. Although steroid-sparing options such as mycophenolate mofetil and cyclosporine are available, studies have shown no treatment benefit for the latter [26] and actually decreased survival for the former [27].
Tacrolimus tends to be the default immunosuppression after transplantation. Its dose is titrated to effect based on whether acute or overlap GVHD develops, but there are reports that sirolimus may be an effective additive. In the initial paper, 8 of 11 patients with sclerotic involvement of their skin responded to therapy with low-therapeutic dosing of sirolimus in combination with their typical immunosuppression [28]. Similarly, in follow-up studies, 70–94 % of patients with sclerotic cGVHD demonstrated some improvement with immunosuppressant regimens inclusive of sirolimus [29, 30]. However, drug interactions, acute kidney injury, and edema may limit its use [31].
Extracorporeal Photopheresis
Extracorporeal photopheresis (ECP) utilizes the same technique as cutaneous photochemotherapy, except that it is performed ex vivo. 8-MOP is incubated with the patient’s leukocytes prior to UVA irradiation, with the end result of increasing graft tolerance and T-cell regulation systemically.
In patients with severe cGVHD, including 20 with extensive cutaneous lichenoid or sclerotic involvement, a 53 % improvement in scoring measurements was seen, and over 80 % of patients were able to decrease their concomitant immunosuppression [32]. Patients with sclerotic-type cGVHD actually may have a slightly higher response rate to ECP (71 %) when compared with all patients with cutaneous cGVHD (59 %) [33]. In a prospective study, patients were able to decrease their standard immunosuppression and showed significant skin improvement in the nonblinded analysis [34].
Rituximab
There is also evidence for the use of rituximab, a monoclonal CD20 chimeric antibody typically reserved for lymphomas, leukemias, rheumatoid arthritis, and autoimmune blistering diseases. It is dosed in the lymphoma protocol, at 375 mg/m2 × 4 weeks, a regimen that can be repeated after 3 months. In early studies of eight patients with sclerotic-type cGVHD, clinical improvement was noted in four, including improvement in pulmonary function testing from loosening of the chest wall [35]. Further prospective studies on cutaneous sclerosis have demonstrated a significant clinical response in 27 % of patients, no higher than patients treated with imatinib [36]. Of note, the study used different validated scoring methods than most cGVHD studies. A meta-analysis of rituximab in this setting asserted that cutaneous response rates vary between 0 % and 83 %, but small study sizes and variable outcome measures prevent clear interpretation [37].
Imatinib
Imatinib, a tyrosine kinase inhibitor targeted to the Philadelphia chromosome, has shown abilities to modulate transforming growth factor–β and platelet-derived growth factor. It is typically dosed at 400 mg daily in adults. Although it demonstrated substantial cutaneous improvement in several early case series, it demonstrated only 30–35 % partial response in larger case series of patients with sclerotic-type cGVHD [38, 39]. When compared prospectively with rituximab, the rate of a significant clinical response in the skin was only 26 % [36].
Ruxolitinib
Ruxolitinib, a selective JAK1/JAK2 inhibitor approved for use in myelofibrosis and polycythemia vera, has recently become a new agent for cGVHD treatment. Overall, the data are sparse. An early preclinical study [40] led to a prospective trial demonstrating an 85 % overall response rate [41], but the response rate differed from rates in previous studies and was based on the ability to taper steroids and systemic immunosuppression by 50 %. No clear specifics were given on the form of cutaneous cGVHD treated.
Related posts:
 Grading and Treatment of Acute Graft-Versus-Host Disease
Grading and Treatment of Acute Graft-Versus-Host Disease
 Wound Care in the Management of Chronic Graft-Versus-Host Disease
Wound Care in the Management of Chronic Graft-Versus-Host Disease
 Clinical Presentation of Acute Cutaneous Graft-Versus-Host Disease
Clinical Presentation of Acute Cutaneous Graft-Versus-Host Disease
 Clinical Presentation of Mucosal Acute and Chronic Graft-Versus-Host Disease
Clinical Presentation of Mucosal Acute and Chronic Graft-Versus-Host Disease
 Diagnosis, Staging, and Treatment of Chronic Graft-Versus-Host Disease
Diagnosis, Staging, and Treatment of Chronic Graft-Versus-Host Disease
 Clinical Presentation of Nonsclerotic Epidermal Chronic Graft-Versus-Host Disease and Hair and Nail Changes
Clinical Presentation of Nonsclerotic Epidermal Chronic Graft-Versus-Host Disease and Hair and Nail Changes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


