Cutaneous Necrotizing Venulitis: Introduction
|
Necrotizing angiitis or vasculitis comprises a diverse group of disorders that combine segmental inflammation with necrosis of the blood vessels. The vascular damage may result from immunologic and/or inflammatory mechanisms. Clinical syndromes are based on criteria that include the gross appearance and the histopathologic alterations of the vascular lesions, the caliber of the affected blood vessels, the frequency of involvement of specific organs, and laboratory abnormalities. Necrotizing vasculitis may be a primary disease, may develop as a feature of a systemic disorder, or may be idiopathic. There is no standard classification of the vasculitides; the American College of Rheumatology classification and the Chapel Hill consensus criteria are widely used.1,2 These classifications are limited in clinical practice, owing to the lack of recognition of certain disorders, the overlap of clinical manifestations, and the lack of precision in the description of cutaneous features.
Necrotizing vasculitis in the skin predominantly involves venules and is known as cutaneous necrotizing venulitis/vasculitis (CNV), cutaneous small-vessel vasculitis, and leukocytoclastic vasculitis. The occurrence of CNV in association with systemic involvement of the small blood vessels has been termed hypersensitivity angiitis/vasculitis, systemic polyangiitis, and microscopic polyangiitis (see Chapter 164). CNV may be restricted to the skin, may occur in association with an underlying chronic disease, may be precipitated by infections or drugs, or may develop for unknown reasons (Table 163-1). Systemic forms of necrotizing vasculitis that affect larger blood vessels are considered in Chapter 164.
|
|
|
Epidemiology
The age of patients has a limited influence on CNV. An associated chronic disorder usually determines the age of the affected individual. CNV is most commonly described in children as Henoch–Schönlein purpura, with an incidence of 20/100,000 individuals less than age 17 in the United Kingdom.3 CNV was the most common type of vasculitis presenting to a dermatology ambulatory department in India.4 Antineutrophil cytoplasmic antibody (ANCA)-associated vasculitides are most common in patients older than 50 years of age. Geographic variations in vasculitis may reflect an environmental influence, and seasonal variations in the incidence of vasculitis may suggest an infectious etiology.5
Etiology and Pathogenesis
Experimental studies in animal models and observations in humans implicate immune complexes as a major pathobiologic mechanism in the production of CNV. Data obtained in animal models suggest that the localization of immune complexes in venules is related to vasoactive amines and subsequent vasopermeability alterations. Additional factors that are operative in the localization of immune complexes include endothelial-cell surface receptors and the defective clearance of immune complexes by the reticuloendothelial system.
The most frequently postulated mechanisms in the production of CNV are the local deposition of circulating immune complexes that are formed during antigen excess or the formation of immune complexes in situ in the skin. Immune complexes may activate the complement system and lead to the generation of C5a anaphylatoxin that degranulates mast cells and attracts neutrophils, which release lysosomal enzymes that damage tissue. The neutrophil superoxide-generating system may produce reactive oxygen products, which also cause tissue injury. The generation of the chemoattractant leukotriene (LT) B4 from infiltrating neutrophils enhances the influx of neutrophils. An infiltrate composed predominantly of neutrophils in the lesional skin of patients with CNV is consistent with tissue damage, which is induced by immune complexes that activate the complement system. The initial neutrophilic process contains few CD3, CD4, CD1a, and CD36 cells whereas these cells are prominent in the later phase, with the adhesion receptors intercellular adhesion molecule-1 (ICAM-1) and lymphocyte function-associated antigen-1 (LFA-1).6 In Henoch–Schönlein purpura, the fragmentation of neutrophils was attributed to apoptosis on the basis of the detection of inducible nitric oxide synthase and nitrotyrosine in the infiltrates and the detection of interleukin-8 (IL-8) on vascular endothelial cells.7 Long penetration PTX3, which inhibits phagocytosis of apoptotic neutrophils by macrophages, was detected in skin biopsy specimens of idiopathic CNV and of Henoch–Schönlein purpura about blood vessels and at sites of infiltrates with leukocytoclasia.8
In CNV, circulating immune complexes have been demonstrated in serum as mixed-type cryoglobulins and indirectly by assays that detect C1q precipitins, materials that bind to complement receptors on human lymphocytoid (Raji) cells, materials that bind to monoclonal rheumatoid factor, and substances that function in the antibody-dependent cellular cytotoxicity inhibition assay. The presence of immune complexes is inferred from the occurrence of serum hypocomplementemia with activation of the classic activating pathway and by the detection of increased plasma levels of C4a and C3a anaphylatoxins.
In CNV, immune complexes have been detected in lesional tissues by their ultrastructural observation as electron-dense subendothelial deposits; the membrane-attack complex, C5b-9, of the complement system has been detected on the surface of endothelial cells and infiltrating neutrophils. Decay-accelerating factor, a regulatory complement protein that prevents the assembly of the membrane-attack complex, was not present on the surface of endothelial cells of the superficial dermal microvasculature. Tissue immune complexes also have been detected by direct immunofluorescence techniques as deposited immunoglobulins and complement proteins. In time–course studies of the evolution of cutaneous vascular lesions, immune reactants have been identified in lesions that are less than 24 hours old. Antigens have been identified only in a few instances as bacterial, viral, mycobacterial, or rickettsial proteins by direct immunofluorescence techniques or by the polymerase chain reaction.
A role for lymphocytes, mononuclear cells, and Langerhans cells in the production of CNV is suggested by a perivenular infiltrate in skin lesions that is rich in lymphocytes with large and hyperchromatic nuclei and by a prominence of other types of mononuclear cells in the vascular skin lesions of patients with CNV and Sjögren syndrome. The lymphocytes express CD3, CD4, CD1a, CD36, ICAM-1, and LFA-1. Lymphocytes may be activated by immune complexes, by cellular immune mechanisms, and by primary activation in autoimmune disease to produce lymphokines. In cell-mediated immune responses in lymphocytic vasculitis, dendritic cells and lymphocytes contribute to the perpetuation of CNV.9 In CNV, there were increased numbers of factor XIIIa+ derived dendrocytes, which are involved in antigen presentation to T cells.10 Endothelial cells also can present antigens to and activate T lymphocytes. Activated macrophages secrete chemokines and lysosomal enzymes. γ/δ T cells have been detected in CNV with a neutrophil-rich pattern and with an infectious etiology.11 In these specimens, a 72-kDa heat shock protein was expressed by endothelial cells and antigen-presenting cells.
The participation of mast cells in CNV is suggested by hypogranulated mast cells with shed extracellular granules and by the development of vascular lesions after the intracutaneous injection of histamine in patients with active episodes of CNV. Mast cells can be activated directly by immune complexes through FcγRIII or by C5a. Through the production of histamine, prostaglandin D2, and cysteinyl LTs, the mast cell could alter venular permeability; interendothelial cell gaps have been noted in venules in patients with CNV. Eosinophils and neutrophils could be recruited by mast cell-derived chemotactic factors. The neutral proteases and acid hydrolases of mast cells may facilitate tissue damage, and the release of tumor necrosis factor-α (TNF-α) may increase expression of E-selectin on endothelial cells and facilitate neutrophil recruitment.
Evidence for the role of the mast cell also is provided by time–course analyses of the sequential histopathologic changes in individuals with physical urticaria. In a patient with circulating immune complexes and hypocomplementemia, in whom cold and trauma elicited CNV, initial mast cell degranulation was followed by the infiltration of neutrophils, the deposition of fibrin, and venular endothelial-cell necrosis. A postulated sequence of events would be the activation of the mast cell by physical stimuli, the release of vasoactive mediators, the deposition of circulating immune complexes with activation of the complement system, the influx of neutrophils, and the development of CNV.12 Another example of the time–course analysis of CNV in human skin was provided by an individual with exercise-induced vasculitis.13 At 3 hours, the number of mast cells decreased, and the eosinophil was the first cell to appear around the venules with the deposition of eosinophil peroxidase. TNF-α levels were elevated, E-selectin was expressed on endothelial cells, and an influx of neutrophils appeared with the deposition of neutrophil elastase and the development of CNV.
Early in the course of necrotizing venulitis, endothelial cells show increased expression of ICAM-1 and E-selectin without the expression of vascular cell adhesion molecule-1 (VCAM-1) in response to TNF-α (see Chapter 162). Because E-selectin is an adhesion molecule for neutrophils and for skin-homing, memory T lymphocytes, the increase in E-selectin is consistent with the appearance of an infiltrate of neutrophils that express CD11b within the first 24 hours.14
In the acute phase of Henoch–Schönein purpura, endothelin-1, serum insulin-like growth factor-1 (IGF-1), IGF-binding protein-3, plasma matrix metalloproteinase-9 (MMP-9), ICAM-1, VCAM-1, TNF- α, IL-I-β, IL-2 receptor, IL-6, IL-8, transforming growth factor-β, vascular endothelial-cell growth factor (VEGF), and urine leukotriene E4 levels were elevated,15–20 and the interferon-γ-inducible protein 10 level was low.21 However, the pathogenic roles of these mediators remain undefined. IgA antiendothelial cell antibodies bind to endothelial cells and enhance endothelial cell production of IL-8 via the mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway.22
In skin biopsy specimens from patients with idiopathic CNV, hypersensitivity vasculitis, urticarial vasculitis, and Henoch–Schönlein purpura, E-selectin was detected on endothelial cells of lesions that were less than 48 hours old and was associated with an infiltrate of neutrophils that expressed CD11b. The endothelial cells expressed human leukocyte antigen DR (HLA-DR) and very late activating antigen-1 but not P-selectin, and the perivascular cells expressed VCAM-1 and HLA-DR. Diminished cutaneous fibrinolytic activity with reduced release of plasminogen activator from venular endothelial cells occurs in patients with CNV; the subsequent reduction in fibrinolytic activity leads to fibrin deposition. Increased levels of plasma thrombomodulin, tissue-type plasminogen activator, and plasminogen activator inhibitor-1 were detected in patients with Henoch–Schönlein purpura.23,24
Cutaneous nerve fibers can release neuropeptides, such as substance P, neurokinin A, and calcitonin gene-related peptide (CGRP), that cause vasodilation (see Chapter 102). Substance P activates mast cells and macrophages and increases fibrinolytic activity that is mediated by plasminogen activator. CGRP induces expression of E-selectin on endothelial cells and is chemotactic for T lymphocytes.
Eosinophils are minor infiltrating cells in CNV except in eosinophilic vasculitis and drug-induced CNV.25 Eosinophils produce LTC4 and platelet-activating factor, which increase vascular permeability. Eosinophil granule proteins are toxic to endothelial cells and cause the release of mediators from mast cells.
Associations have been recognized between small-vessel necrotizing vasculitis and ANCAs, which have specificity for proteins of the cytoplasmic granules of neutrophils and the lysosomes of monocytes. Two forms are recognized: (1) cytoplasmic (c-ANCAs) that are directed against proteinase 3(PR3) and (2) perinuclear (p-ANCAs) that are directed against myeloperoxidase (MPO). TNF-α facilitates neutrophil activation and cell surface expression of PR3 and MPO, which bind to ANCAs and increase the adherence of neutrophils to endothelial cells resulting in endothelial cell injury. Antiendothelial-cell antibodies have been detected in the sera of patients with systemic vasculitis, rheumatoid arthritis with vasculitis, microscopic polyangiitis, and Sneddon syndrome.
An increased prevalence of the HLA haplotype HLA-A11, Bw35 in patients with CNV and associated connective-tissue disorders suggests that genetic factors may be operative. HLA-DRB1 genotype associations were detected in patients with CNV and Henoch–Schönlein purpura in Spain.26
Clinical Findings
The skin lesions of CNV are polymorphous; however, erythematous papules that do not blanch when the skin is pressed, which are known as palpable purpura, are the signature lesions (Fig. 163-1A). Macules, papules, urticaria/angioedema, pustules, vesicles, hemorrhagic blisters (Fig. 163-1B), necrosis and ulcers (Fig. 163-1C), and livedo reticularis may be present. Occasionally, there is subcutaneous edema below the area of the dermal lesions.
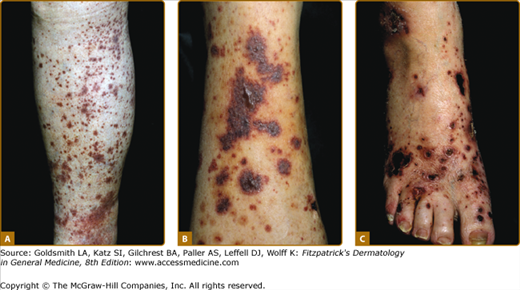
The eruption most often appears on the lower extremities or over dependent areas, such as the back and gluteal regions. The lesions may occur anywhere on the skin but are uncommon on the face, palms, soles, and mucous membranes. The clinical lesions are episodic and may recur over weeks to years. Palpable purpura persists for 1 to 4 weeks and resolves at times with transient hyperpigmentation and/or atrophic scars. Lesional symptoms include pruritus or burning and, less commonly, pain.
An episode of cutaneous vascular lesions may be attended by fever, malaise, arthralgias, or myalgias irrespective of a defined underlying or associated disease. Systemic involvement of the small blood vessels most commonly occurs in the synovia, gastrointestinal tract, voluntary muscles, peripheral nerves, and kidneys.
CNV is associated with connective-tissue diseases, notably rheumatoid arthritis, Sjögren syndrome, systemic lupus erythematosus (SLE), and hypergammaglobulinemic purpura. It rarely occurs in mixed connective-tissue disease, relapsing polychondritis, and scleroderma. In patients with rheumatoid arthritis and CNV, the development of vascular lesions is related to the severity of the disease, which is generally but not always seropositive, and the presence of anti-Ro antibodies. Subcutaneous nodules and cutaneous ulcers may be present. Patients with rheumatoid arthritis often have involvement of larger vessels with associated peripheral neuropathy, nail-fold infarcts, and digital gangrene.
In patients with SLE, CNV is a frequent manifestation27 and is associated with exacerbations of the underlying disease. Patients with anti-Ro antibody have a greater risk for the development of CNV, with an Odds ratio 1.63.28 Vasculitis, however, is rare in patients with subacute cutaneous lupus erythematosus. Some women with necrotizing vasculitis without connective-tissue disease have anti-Ro antibodies, and their infants may be born with neonatal lupus erythematosus.
In patients with Sjögren syndrome, the vascular lesions are located predominantly on the lower extremities and appear after exercise. Both hyperpigmentation and cutaneous ulcers are common features. Patients with Sjögren syndrome and CNV have a higher prevalence of articular involvement, peripheral neuropathy, Raynaud phenomenon, and renal involvement29 as well as the presence of anti-Ro antibody. Hypergammaglobulinemic purpura occurs in older women and may be associated with Sjögren syndrome, SLE, or a lymphoproliferative disorder. Dermatomyositis in children, but not in adults, may be associated with vasculitis of the gastrointestinal tract.
Paraneoplastic vasculitis describes CNV with associated malignant conditions, which include Hodgkin lymphoma, lymphosarcoma, adult T-cell leukemia, mycosis fungoides, myelofibrosis, acute and chronic myelogenous forms of leukemia, B-cell chronic lymphocytic leukemia, multiple myeloma, IgA myeloma, diffuse large-cell leukemia, hairy-cell leukemia, squamous-cell bronchiogenic carcinoma, adenocarcinoma of the lung, prostatic carcinoma, renal carcinoma, bladder carcinoma, and colon carcinoma.30 However, the association between CNV and neoplasia is rare. It is not necessary to evaluate all patients with CNV for associated malignant conditions.
Cryoglobulins (see Chapter 169), especially mixed types II and III, may be found in patients with idiopathic CNV and in patients with CNV that is associated with connective-tissue diseases, lymphoproliferative disorders, and hepatitis A, B, and C virus infections. Hepatitis C virus is the most common infection, especially when it is associated with cryoglobulinemia.31,32 CNV occurs in patients with cystic fibrosis, inflammatory bowel diseases of the colon, and Behçet disease.33
ANCAs are associated with various forms of necrotizing vasculitis. ANCAs are present in patients with microscopic polyangiitis and cutaneous vasculitis with hepatitis C virus infection (see Chapter 164). The most common cutaneous feature in patients with ANCAs is palpable purpura. Microscopic polyangiitis is associated with small-vessel systemic vasculitis that involves the cutaneous venules and arterioles, the kidneys with necrotizing and crescentic glomerulonephritis, the lungs with pulmonary hemorrhage or interstitial pneumonia, and p-ANCAs. Erythematous macules, purpura, and livedo reticularis are cutaneous manifestations in microscopic polyangiitis.34,35 A male patient has experienced microscopic polyangiitis restricted to the skin and p-ANCAs for 20 years without progression to systemic vascular disease.36 IgA ANCAs are present in acute Henoch–Schönlein purpura in children.
Anticardiolipin antibodies occur in patients with various forms of necrotizing vasculitis. IgA cardiolipin antibodies of the IgA class were detected in 6 of 10 patients with idiopathic CNV.37 IgA anticardiolipin and IgA and IgM antiphosphatidyl serine–prothrombin complex antibody levels are elevated in adults with Henoch–Schönlein purpura and CNV that is restricted to the skin.38 IgG and IgM anticardiolipin antibodies have been detected in some individuals with livedoid vasculitis.39
Infections and drugs may precipitate episodes of CNV. The most commonly recognized infectious agents are β-hemolytic Streptococcus, Staphylococcus aureus, Mycobacterium leprae, and hepatitis B and C viruses. Transient episodes of urticaria may occur early in the course of hepatitis B virus infection and represent immune complex-induced vasculitis; episodes of palpable purpura may occur in patients with chronic active hepatitis. Cutaneous vasculitis has been recognized in a limited number of individuals with human immunodeficiency virus infection; the skin lesions consisted of palpable purpura, which at times had a follicular localization, and cutaneous ulcers.
Erythema nodosum leprosum (see Chapter 186), which appears as cutaneous nodules in lepromatous leprosy, is a form of necrotizing vasculitis that involves capillaries, venules, arterioles, small-to-medium-sized arteries, and veins. The vascular lesions occur spontaneously or are precipitated by the administration of chemotherapeutic agents. They may be accompanied by fever, malaise, arthralgias, lymphadenopathy, and polyneuritis.
Necrotizing vasculitis caused by the direct invasion of the blood-vessel wall occurs in septicemia due to Neisseria meningitidis, Neisseria gonorrhoeae, Pseudomonas, Hemophilus influenzae, rickettsia, candida, and infectious endocarditis; in Rocky Mountain spotted fever; and in infections localized at the site of a catheter.
Palpable purpura is one of the less common forms of drug reactions (see Chapter 41). The most commonly incriminated therapeutic agents were propylthiouracil, hydralazine, granulocyte colony-stimulating factor (G-CSF)/granulocyte-macrophage colony-stimulating factor (GM-CSF), allopurinol, cephaclor, minocycline, penicillamine, phenytoin, isotretinoin, and methotrexate in a Medline database search from 1965 to 1999.40 Propylthiouracil41
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


