Introduction354
INTRODUCTION
Disease
Histological features
Generalized myxedema
Subtle changes: mucin deposition often only perivascular or perifollicular; no fibroblast changes
Pretibial myxedema
Increased mucin often localized to mid and lower dermis; fibroblasts sometimes stellate, but not increased
Scleredema
Thickening of reticular dermis due to swelling and separation of collagen bundles; no significant hyperplasia of fibroblasts; variable interstitial mucin, sometimes minimal in late stages; no deep inflammatory infiltrate as in morphea
Scleromyxedema
Prominent fibroblastic proliferation and increased collagen; variable mucin increase
Papular mucinosis
Discrete form resembles focal mucinosis; slight proliferation of fibroblasts in generalized form
Acral persistent papular mucinosis
Resembles papular mucinosis but mucin deposition and fibroblast proliferation less pronounced
Cutaneous mucinosis of infancy
May be a variant of scleromyxedema
Nephrogenic systemic fibrosis
Resembles scleromyxedema but usually has subcutis involvement; less mucin; no plasma cells
Reticular erythematous mucinosis
Superficial and characteristic mid-dermal perivascular lymphocytic infiltrate; sometimes deeper extension around eccrine coils; mucin usually prominent
Focal mucinosis
Dome-shaped solitary nodule with prominent mucin in upper or entire dermis with variable collagen replacement
Digital mucous cyst
Mucinous pool with stellate fibroblasts resembling focal mucinosis, or a cavity with a myxoid connective tissue wall
Mucocele
Pseudocystic space with surrounding macrophages and vascular loose fibrous tissue or granulation tissue with mucin, muciphages, and inflammatory cells
Nevus mucinosus
Mucin in expanded papillary dermis; few fibroblasts present
Secondary dermal mucinoses
Mucin and changes of underlying disease such as lupus erythematosus, Degos’ disease, Jessner’s lymphocytic infiltrate, granuloma annulare, and dermatomyositis
Follicular mucinosis (alopecia mucinosa)
Mucin in hair follicles and attached sebaceous gland with some dissolution of cellular attachments; variable inflammatory infiltrate; infiltrate is dense and atypical in secondary follicular mucinosis complicating lymphoma or mycosis fungoides
Mucopolysaccharidoses
Metachromatic granules in fibroblasts and sometimes eccrine glands and keratinocytes; extracellular mucin in maculopapular lesions of Hunter’s syndrome
DERMAL MUCINOSES
GENERALIZED MYXEDEMA
Histopathology
PRETIBIAL MYXEDEMA (THYROID DERMOPATHY)
Histopathology
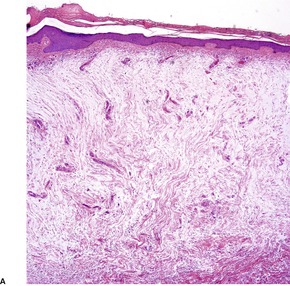
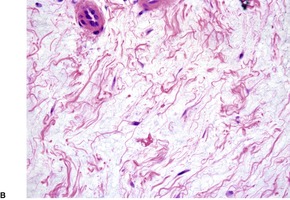
Fig. 13.1
Fig. 13.2
PAPULAR MUCINOSIS AND SCLEROMYXEDEMA
Histopathology
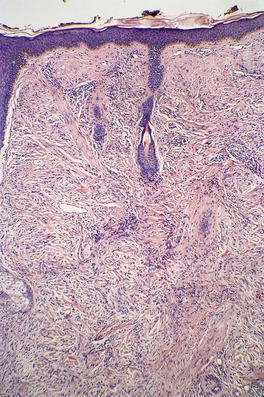
Fig. 13.3
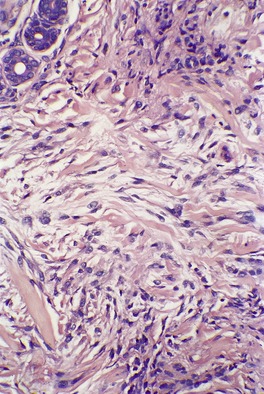
Fig. 13.4
ACRAL PERSISTENT PAPULAR MUCINOSIS
Histopathology
Electron microscopy
CUTANEOUS MUCINOSIS OF INFANCY
SELF-HEALING JUVENILE CUTANEOUS MUCINOSIS
Histopathology
NEPHROGENIC SYSTEMIC FIBROSIS
Histopathology
RETICULAR ERYTHEMATOUS MUCINOSIS (REM)
Histopathology
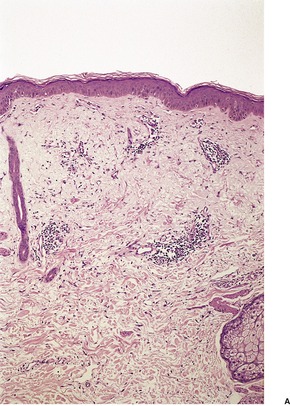
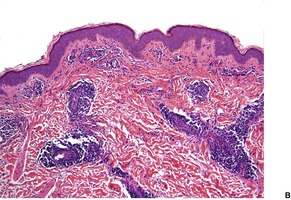
Fig. 13.5
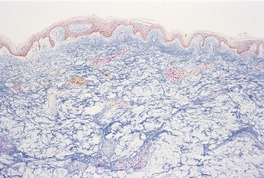
Fig. 13.6
SCLEREDEMA
Histopathology241. and 256.
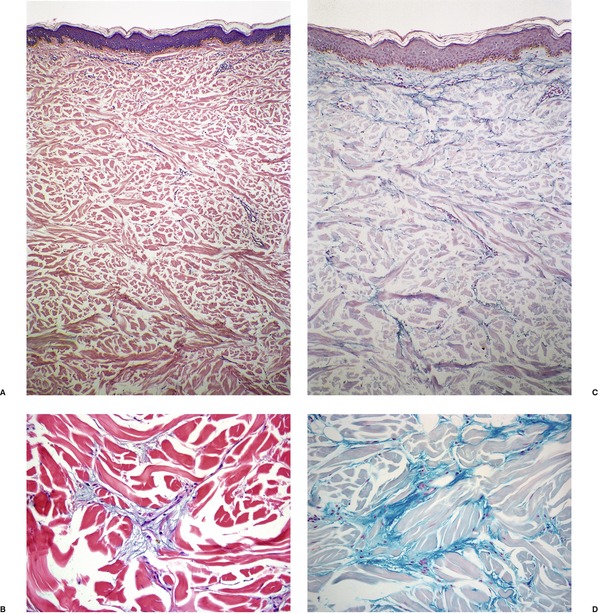
Fig. 13.7
FOCAL MUCINOSIS
Histopathology291
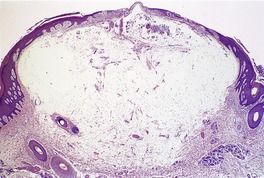
Fig. 13.8
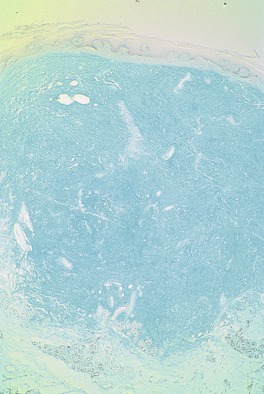
Fig. 13.9
Electron microscopy
DIGITAL MUCOUS (MYXOID) CYST
Histopathology302
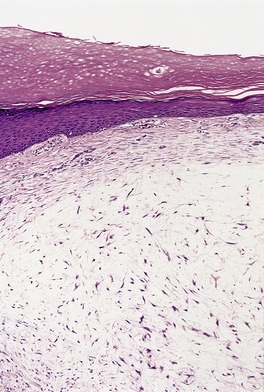
Fig. 13.10
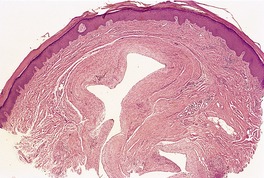
Fig. 13.11
MUCOCELE OF THE LIP
Histopathology
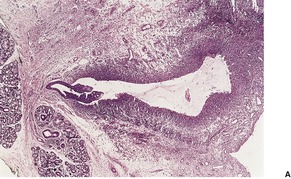
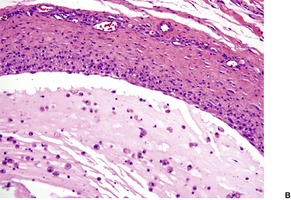
Fig. 13.12
CUTANEOUS MYXOMA AND CARNEY’S COMPLEX
Histopathology
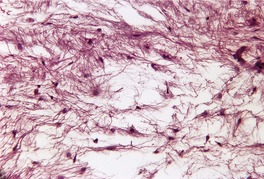
Fig. 13.13
NEVUS MUCINOSUS (MUCINOUS NEVUS)
Histopathology
PROGRESSIVE MUCINOUS HISTIOCYTOSIS
SECONDARY DERMAL MUCINOSES
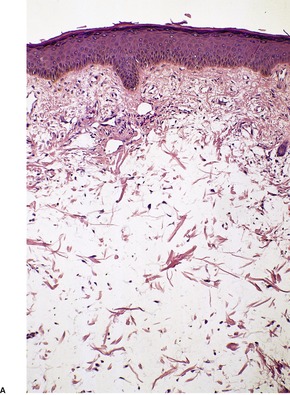
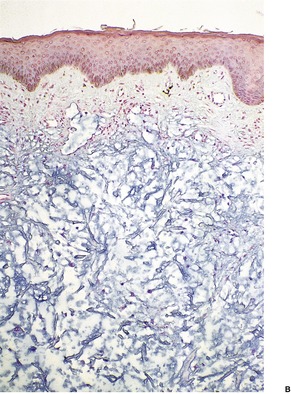
Fig. 13.14
FOLLICULAR MUCINOSES
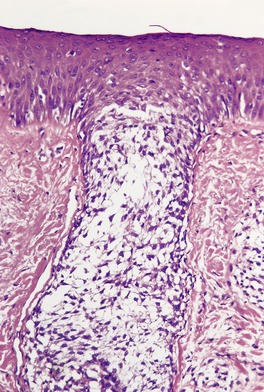
Fig. 13.15
FOLLICULAR MUCINOSIS (ALOPECIA MUCINOSA)
Histopathology362.405. and 406.
Fig. 13.16
SECONDARY FOLLICULAR MUCINOSES
PERIFOLLICULAR MUCINOSIS
EPITHELIAL MUCINOSES
MUCOPOLYSACCHARIDOSES
Histopathology
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



Cutaneous mucinoses
Dermal mucinoses355
Generalized myxedema355
Pretibial myxedema (thyroid dermopathy)355
Papular mucinosis and scleromyxedema355
Acral persistent papular mucinosis357
Cutaneous mucinosis of infancy357
Self-healing juvenile cutaneous mucinosis358
Nephrogenic systemic fibrosis358
Reticular erythematous mucinosis (REM)358
Scleredema359
Focal mucinosis360
Digital mucous (myxoid) cyst360
Mucocele of the lip362
Cutaneous myxoma and Carney’s complex363
Nevus mucinosus (mucinous nevus)364
Progressive mucinous histiocytosis364
Secondary dermal mucinoses364
Epithelial mucinoses367
Mucopolysaccharidoses367
The mucinoses are a diverse group of disorders which have in common the deposition of basophilic, finely granular and stringy material (mucin) in the connective tissues of the dermis (dermal mucinoses),1. and 2. in the pilosebaceous follicles (follicular mucinoses), or in the epidermis and tumors derived therefrom (epithelial mucinoses). 3 The most important mucinoses are the dermal ones where glycosaminoglycans (GAGs), also known as acid mucopolysaccharides, accumulate in the dermis.4. and 5. Glycosaminoglycans are a major class of extracellular complex polysaccharides. 6 There are four classes:
• heparin/heparan
• chondroitin/dermatan
• keratan
• hyaluronan (hyaluronic acid).
Hyaluronan (HA) and heparin sulfate comprise the major fraction of the vertebrate pericellular matrix. 7 The total amount of HA in an adult is estimated to be 20 g and turnover is about 15 g/day. 7 It is found in the vitreous humor of the eye, the extracellular matrix, and the intercellular space of the epidermis where it appears to be involved in cell–cell interactions. CD44 is thought to be the primary receptor for HA on cells. 7
Hyaluronan, in contrast to the other GAGs, is non-sulfated and is synthesized, not on the Golgi apparatus, but on the inner face of the plasma membrane of the fibroblast.7. and 8. Three hyaluronan synthases (HAS) are involved in its synthesis. It is removed by lymphatics and degraded in the liver and lymph nodes. Polymer size confers specific functions on HA which include wound healing, angiogenesis, aspects of tumor growth, and host–pathogen interactions. 7
Hyaluronan is highly hydrophilic and water drawn into the matrix creates a turgor that enables it to withstand compressive forces.5. and 9. The loss of this water during paraffin processing results in residual basophilic strands and granules in widened dermal spaces in sections stained with hematoxylin and eosin. Hyaluron is poorly fixed by 10% formalin and some is also lost during tissue processing.
Fragmentation of the dermal collagen is quite common in the dermal mucinoses. Collagen type III is often reduced although there is a compensatory increase in type I collagen. 10
The pathogenetic mechanisms involved in the accumulation of mucin in the skin are poorly understood. In many of the mucinoses there appears to be an increased production of acid mucopolysaccharides by fibroblasts, although in myxedema it has been suggested that impaired degradation leads to accumulation of mucin in the dermis (see below). Several of the cutaneous mucinoses have been reported in patients with HIV infection, although only the association with papular mucinosis (lichen myxedematosus) seems to be statistically significant. 11 The relationship between the infection and the mucin deposition is unclear. 11
In the mucopolysaccharidoses, which are best considered separately from the other mucinoses, the predominant dermal mucin is chondroitin sulfate, rather than hyaluronan.
The two most common methods used for demonstrating mucin in the skin are the Alcian blue technique at pH 2.5 and the colloidal iron stain, with which acid mucopolysaccharides are blue-green. Metachromasia of mucin is usually demonstrated with the toluidine blue or Giemsa methods. It has been suggested that fixation in a 1% solution of cetylpyridinium chloride in formalin, followed by colloidal iron staining of the paraffin sections, gives the best definition of glycosaminoglycans in the skin. 12
The histological features of the various mucinoses are summarized in Table 13.1.
The distribution of the glycosaminoglycans is said to differ in the various dermal mucinoses. However, in a comparative study some years ago, Matsuoka and colleagues found that the distribution of these substances is generally not diagnostically specific. 12 Accordingly, clinicopathological correlation is important in this group. Scleredema and scleromyxedema differ from the other dermal mucinoses by the presence of collagen deposition and fibroblast hypertrophy and/or hyperplasia, in addition to the deposition of mucin.
Alajlan and Ackerman have questioned the legitimacy of the concept of the dermal mucinoses. 13 They claim that the term has not yet been defined meaningfully and that each of the ‘so-called dermal mucinoses can be identified on the basis of distinctive clinical and histopathological features’. 13
The case of cutis verticis gyrata caused by the deposition of mucin in the dermis was not associated with any of the mucinoses described elsewhere, so it is mentioned here for completeness. 14
Myxedema is one of several cutaneous changes in hypothyroidism.4.15.16. and 17. The changes are most pronounced around the eyes, nose, and cheeks, often giving a characteristic facies, and also on the distal extremities. The skin is cold, dry, and pale with widespread xerosis, especially on the extensor surfaces. 18 A diffuse, non-scarring alopecia may be present. 19 Cutis verticis gyrata is a rare presentation. 20
Palmoplantar keratoderma is a poorly recognized presentation of myxedema. 21 Glycosaminoglycans are deposited in other organs of the body as well as the skin and it has been suggested that there is impaired degradation, rather than increased synthesis, of these substances.5. and 22. Muscle weakness is sometimes present. 23
Generalized and pretibial myxedema have been grouped together as ‘dysthyroidotic mucinoses’ in the excellent review of the cutaneous mucinoses by Rongioletti and Rebora. 24
In most cases, the changes are subtle with only small amounts of mucin in the dermis. 5 This is predominantly hyaluronic acid. Sometimes this material is deposited only focally around vessels and hair follicles. 5 There may be mild hyperkeratosis and keratotic follicular plugging, reflecting the xerosis that is present clinically. Elastic fibers are sometimes fragmented and reduced in amount.
Pretibial myxedema (thyroid dermopathy) is found in 1–4% of patients with Graves’ disease, particularly those with exophthalmos, but it may not develop until after the correction of the hyperthyroidism. 25 Rarely, it may precede the diagnosis of hyperthyroidism. 26 It may also occur in patients with non-thyrotoxic Graves’ disease27 and occasionally in association with Hashimoto’s thyroiditis. 28 It has been associated with an ectopic, hyperplastic nodule arising in a thyroglossal duct residue. 29 Pretibial myxedema has been reported in euthyroid patients with stasis dermatitis, 30 and in patients with morbid obesity who have lower leg lymphedema. 31
It presents as sharply circumscribed nodular lesions, diffuse non-pitting edema, or elephantiasis-like thickening of the skin.18.32. and 33. There may be overlap of these lesions or progression from nodular to more diffuse plaques. 25 The anterior aspect of the lower legs, sometimes with spread to the dorsum of the feet, is the most usual site of involvement, although, rarely, the upper trunk, upper extremities, and even the face, neck or ears have been involved.34.35. and 36. Edema of the eyelids, not associated with the deposition of mucin, may occur in hyperthyroidism. 37 Localization to scar tissue, skin grafts, and the toes has been reported.38.39.40.41.42. and 43. Acropachy (digital clubbing and diaphysial proliferation) is rare, with an incidence of less than 1%. 44 Because sites other than the pretibial region can be affected, the term thyroid dermopathy is more appropriate. 18 Slow resolution of the lesions often occurs after many years. Hyperhidrosis and/or hypertrichosis, limited to areas of thyroid dermopathy, have been reported.18. and 45.
The precise pathogenesis of pretibial myxedema is unknown. The theory that pretibial myxedema results from the stimulation of fibroblasts by LATS (long-acting thyroid stimulator) is no longer tenable, although one or multiple other substances or autoantibodies are presumably involved.34. and 46. A circulating factor that stimulates increased synthesis of glycosaminoglycans by normal skin fibroblasts is present in increased amounts in patients with pretibial myxedema.47. and 48. Furthermore, fibroblasts from pretibial skin cultured in the presence of serum from patients with pretibial myxedema produce increased amounts of hyaluronic acid. 49 The response of refractory cases of pretibial myxedema to octreotide (a somatostatin analogue with insulin-like growth factor 1 (IGF-1) antagonist properties) suggests that expression of IGF-1 receptor on fibroblasts may be up-regulated in this condition, leading to increased secretion of hyaluronic acid. 50 Other explanations for this effect are possible. There is no primary lymphatic abnormality. 25 Hydrostatic forces appear to play a role in the localization of the pretibial mucinosis. 51
There are large amounts of mucin deposited in the dermis, particularly in the mid and lower thirds (Fig. 13.1 and Fig. 13.2). Initial deposition is often in the papillary dermis with subsequent extension into the deeper tissues. 18 This manifests as basophilic threads and granular material with wide separation of collagen bundles. 4 There is no increase in fibroblasts although a few stellate forms may be present. There may be overlying hyperkeratosis which can be quite marked in clinically verrucous lesions. 52 A mild superficial perivascular chronic inflammatory cell infiltrate is often present. 22 In patients with underlying stasis dermatitis, the deposition of mucin is within the papillary dermis with sparing of the reticular dermis. 30 Angioplasia and hemosiderin deposition are additional features in the dermis.
Pretibial myxedema. (A) There is pallor of the dermis and (B) the collagen bundles in the dermis are widely spaced, a consequence of the increased amount of interstitial mucin. (H & E)
Pretibial myxedema. There are large amounts of dermal mucin. (Alcian blue)
Elastic tissue stains show fragmentation and a reduction in elastic tissue, a finding confirmed on electron microscopy53 which also shows microfibrils with knobs (glycosaminoglycans) 54 or amorphous material (glycoproteins) 55 on the surface of fibroblasts that have dilated endoplasmic reticulum.54. and 55.
Papular mucinosis (lichen myxedematosus) is a rare cutaneous mucinosis in which multiple, asymptomatic, pale or waxy papules, 2–3 mm in diameter, develop on the hands, forearms, face, neck, and upper trunk. 56 A discrete papular variant also occurs. 57 It has been reported, rarely, in patients with hepatitis C58 and with HIV infection11.59.60.61.62. and 63. and also in the L-tryptophan-induced eosinophilia–myalgia syndrome, 64 in generalized morphea, 65 subclinical hypothyroidism, 66 and in patients with morbid obesity. 67 Self-healing cases have been reported, 68 indicating the overlap between this entity and self-healing cutaneous mucinosis (see below). Papular mucinosis with systemic involvement has also been reported. 69
Scleromyxedema is a variant in which lichenoid papules and plaques are accompanied by skin thickening involving almost the entire body.70.71.72. and 73. The face, neck, hands, and forearms are sites of predilection. Involvement of the glabella region may give rise to bovine or leonine facies.70. and 74. The Koebner phenomenon due to a scratch test has been reported. 75 Adults in the third to fifth decades, and older, are most commonly affected. A paraproteinemia, particularly of IgG lambda type, is almost invariably present in scleromyxedema and sometimes in papular mucinosis.4.76. and 77. Other classes of immunoglobulins are sometimes present; 78 a few patients have a normal immunoglobulin profile. 79 Multiple myeloma and Waldenström’s macroglobulinemia are rare associations.80. and 81. Bizarre neurological symptoms, 82 underlying carcinoma,56.83. and 84. chronic hepatitis C, 85 pachydermoperiostosis, 56 dermatomyositis, 86 scleroderma,81. and 87. atherosclerosis, 88 esophageal aperistalsis, 89 and multiple keratoacanthomas78 have all been reported in association with scleromyxedema. 81
Scleromyxedema is usually progressive, but spontaneous resolution has been reported. 90 Mucin has been noted in other organs in a few autopsy cases, but it has been specifically excluded in others.56.91.92. and 93. Rarely, hypothyroidism has been present.94. and 95.
In 2001, Rongioletti and Rebora provided an updated classification of papular mucinosis/lichen myxedematosus/scleromyxedema. 96 The generalized and sclerodermoid form is best known as scleromyxedema. The localized form has several subtypes. 97 They include acral persistent papular mucinosis, self-healing papular mucinosis (juvenile and adult variants), papular mucinosis of infancy, and nodular lichen myxedematosus as variants, despite the absence of fibrosis in some of these conditions.
It has been postulated that a serum factor stimulates fibroblast proliferation and increased production of glycosaminoglycans.87. and 98. Whether this factor is identical to the monoclonal immunoglobulin is controversial.81. and 99. In one study, cultured skin fibroblasts from a patient with scleromyxedema produced an IgG immunoglobulin. 100
An unusual scleromyxedema-like disease has been reported in renal dialysis patients and in other circumstances. The terms ‘nephrogenic fibrosing dermopathy’ and ‘nephrogenic systemic fibrosis’ have been applied to this condition. It is considered, in detail, below.
Response to high- and low-dose intravenous immunoglobulin, interferon alfa, prednisone, retinoids, high-dose pulsed methylprednisolone, thalidomide, melphalan, electron-beam therapy, extracorporeal photopheresis, and autologous stem cell transplantation has been reported.101.102.103.104.105.106.107.108.109.110.111.112.113.114.115.116. and 117. The localized type has been treated with tacrolimus ointment. 97
The histopathological features of scleromyxedema are the most precise of any of the mucinoses.22. and 70. In addition to the dermal deposits of mucin, there is a marked proliferation of fibroblasts and increased collagen deposition in the upper and mid dermis (Fig. 13.3 and Fig. 13.4). 22 The fibroblasts are irregularly arranged and the collagen, which is most pronounced in older lesions, has a whorled pattern. Flattening of the epidermis and atrophy of pilosebaceous follicles are secondary changes. Sweat duct proliferation is rare. 118 Elastic fibers are often fragmented. 100 A sparse perivascular infiltrate of lymphocytes is often present. Occasionally, eosinophils89. and 91. or mast cells119 are prominent. In one case there was a prominent perivascular infiltrate of plasma cells which were monotypic for λ light chain. 120
Scleromyxedema. The dermis contains an increase in fibroblasts, collagen, and interstitial mucin. (H & E)
Scleromyxedema. The characteristic triad of an increase in fibroblasts, collagen, and mucin is present. (H & E)
An unusual granulomatous variant of scleromyxedema has been reported. There were interstitial granuloma annulare-like features. 121
The changes in nephrogenic systemic fibrosis are similar to those seen in scleromyxedema (see below). Involvement of the subcutis is a common feature of nephrogenic systemic fibrosis and rare in scleromyxedema.
Ultrastructurally, the fibroblasts have prominent rough endoplasmic reticulum. Proteoglycans are present between the collagen bundles. 122
The changes in papular mucinosis are not as characteristic. In the discrete form, the changes may be indistinguishable from focal mucinosis, 123 although in the more generalized cases a slight proliferation of fibroblasts is often present in addition to the mucin deposition in the upper dermis. 94 Sclerotic features are absent. 67
Acral persistent papular mucinosis, which affects mainly women, is characterized by discrete, flesh-colored papules 2–5 mm in diameter on the back of the hands and, less commonly, on the forearms124.125.126.127.128.129.130. and 131. and calves. 132 Approximately 30 cases have been reported. 133 The lesions increase in number with the years. There is usually no associated disease. 134 It is best regarded as a variant of papular mucinosis (lichen myxedematosus).96. and 135.
A patient with a self-healing plaque on the dorsum of one hand has been reported as self-healing localized cutaneous mucinosis. 136 This case does not fit neatly into any of the current classifications of the mucinoses.
The lesions in acral persistent papular mucinosis are asymptomatic. No treatment is effective. 137
This condition shares some histological features with papular mucinosis (see above) by having mucin deposition and a proliferation of fibroblasts. However, the mucin is usually confined to the upper and mid dermis, in contrast to the more widespread distribution in the dermis in papular mucinosis. A spared grenz zone is usually present. 135 Furthermore, fibroblastic proliferation is not as pronounced as in papular mucinosis; it may be absent in early cases. 131
Altered fibroblasts with concentric lysosomal structures in their cytoplasm have been reported. 127
There have been several reports of infants presenting with multiple, small, papular lesions on the upper extremities or trunk.2.138.139.140. and 141. The distribution of the lesions and their early onset raise the possibility that the lesions reported are connective tissue nevi of proteoglycan type (nevus mucinosus).142. and 143. There has been abundant mucin in the papillary dermis, no significant increase in fibroblasts, and a few chronic inflammatory cells in a perivascular distribution. In one case, however, there was both fibrosis and fibroblast proliferation, leading the authors to suggest that cutaneous mucinosis of infancy could be a pediatric form of papular mucinosis (lichen myxedematosus). 144 This is the currently accepted view.
There have been several reports of a cutaneous mucinosis characterized by the rapid onset in childhood of infiltrated plaques on the head and torso and deep nodules on the face and periarticular region, with spontaneous resolution in weeks or months.145.146.147.148.149. and 150. Other reports have described a familial variant, 151 and rare cases in adults.152. and 153. The existence of self-healing cases of papular mucinosis (lichen myxedematosus) 68 illustrates the arbitrary nature of the current classification of some mucinoses and lends support to the notion of Rongioletti and Rebora in 2001 that many of these entities are clinical variants of papular mucinosis (lichen myxedematosus).96. and 154. Notwithstanding this view, this mucinosis is thought to represent a reactive or reparative response to some antigenic stimulation, such as inflammation or a viral infection. 155 It has developed in a child with a nephroblastoma who was undergoing chemotherapy. 149
Biopsies of this condition have shown a normal epidermis overlying an ‘edematous’ dermis resulting from mucin separating collagen bundles in the dermis. An increase in fibroblasts has been reported. 149 A sparse perivascular inflammatory cell infiltrate is sometimes present and CD68-positive macrophages are present in the base of the mucin deposits. 156
The subcutaneous nodules are composed of large mucinous lobules (islands) separated by fibrous tissue bands containing a sparse population of fibroblast-like spindled cells. 155 A sparse lymphocytic infiltrate may be present. The mucinous lobules, which stain strongly for Alcian blue at pH 2.5, contain a lobular array of stellate, spindled, and in some cases rhabdoid or gangliocyte-like giant cells.155. and 157. There is some resemblance to proliferative fasciitis. 157 The dermis overlying these subcutaneous nodules usually shows a mild increase in mucin but no increase in fibroblasts. Stains for CD34, smooth muscle actin, muscle specific actin, cytokeratins, and epithelial membrane antigen are negative. 155
This condition, previously called nephrogenic fibrosing dermopathy, is a scleromyxedema-like disease of the skin and other organs that occurs in a subset of patients with renal insufficiency.158.159.160. and 161. Renal dialysis is not a prerequisite for its development. 162 It may follow severe acute-kidney injury. 163 Cowper has done much of the work in describing the features of this entity.160.164.165. and 166.
It evolves over a period of days to weeks with thickening and hardening of the skin, muscle weakness, and generalized pain. Diaphragmatic involvement is a serious complication that can lead to death.167.168. and 169. The skin lesions are indurated papules and plaques, involving predominantly the trunk and extremities.170. and 171. Facial papules are rare. 172 A patient with lesions limited to the breast, mimicking inflammatory breast carcinoma, has been reported. 173 In another case there was a localized plaque on the forearm along a vein that was traumatized during the infusion of erythropoietin. 174 This substance has been suggested as a possible cofactor in producing this disease. 175 Pediatric cases are rare. 176
The sudden appearance of this condition in 1997 and its association with renal insufficiency led to an extensive search for the etiological agent. In 2006, gadolinium-based contrast agents used in magnetic resonance imaging (MRI) were implicated as the causative agent, in patients who had renal disease and were often acidotic. 177 Gadolinium had been used a few weeks prior to the onset of the disease in each case.178.179.180.181.182. and 183. Gadolinium has since been identified in the tissues of affected patients.160.184.185.186. and 187. It has recently been suggested that the gadolinium product involved is gadodiamide. 188 Gadodiamide is retained in apatite-like deposits. It may be mobilized over time from these bone stores. 189 The fibrosis caused by this agent may result from the up-regulation of transglutaminases in the affected tissue. 190 Transforming growth factor beta (TGF-β) also plays a role in producing the fibrosis. 191
Parenthetically, the author has never seen a case, possibly because the dose of gadolinium used in Brisbane is lower than in the United States. Nephrogenic systemic fibrosis has recently been reported in a gadolinium-naive renal transplant recipient. 192
Improvement in cutaneous lesions has been reported with UVA1 phototherapy, 193 high-dose intravenous immunoglobulin, 194 and extracorporeal photopheresis.188. and 195. Retinoids have been used with PUVA therapy, giving significant clinical improvement. 196
Hopefully this disease is destined to become another historical footnote, like the eosinophilia–myalgia syndrome and others. 197
The dermis and septa of the subcutaneous fat are thickened by dense collagen bundles in haphazard array. There is a proliferation of plump spindle-shaped cells and variable interstitial mucin.198.199.200.201. and 202. Histiocytes positive for CD68, and dendrocytes that are positive for factor XIIIa are also present. 203 Many of the spindle-shaped cells show dual immunoreactivity for CD34 and procollagen.204.205. and 206. Small multinucleate histiocytes have also been described. 199 Various elastic tissue changes have been reported, including the presence of elongated fibers, 202 calcification of elastic fibers resembling pseudoxanthoma elasticum, 207 and the rare development of generalized elastolysis. 208 Calciphylaxis, metastatic calcification,209. and 210. and osseous metaplasia uncommonly develop. 211 Osteoclast-like giant cells and subcutaneous fat necrosis have also been reported,209. and 212. as has a septal panniculitis mimicking erythema nodosum. 213 Hyaluronan, a mucin that is poorly retained in formalin-fixed material, can be demonstrated in the papillary dermis by special techniques, 214 and cultured fibroblasts from lesional skin also produce increased amounts of this mucin. 215
While one study failed to find any histological differences from scleromyxedema, 216 the presence of subcutaneous involvement in nephrogenic systemic fibrosis is a distinguishing feature. 217 Mucin deposition is less, 218 but this may, in part, be related to an increased proportion of poorly retained hyaluronan. Plasma cells are usually not present in this condition, but small clusters may be present in scleromyxedema. 218
Reticular erythematous mucinosis (REM) was first described by Steigleder and colleagues in 1974. 219 It presents with erythematous maculopapules and infiltrated plaques, often with a reticulated or net-like pattern, in the midline of the back or chest, sometimes spreading to the upper abdomen.220.221.222.223. and 224. Rarely, the face and arms225 are involved and one case is said to have involved the gums. 226 There is a predilection for young to middle-aged females. Sunlight and hormonal influences may cause exacerbations and induce mild pruritus.4.224. and 227. The lesions may subside after many years. Progression to cutaneous lupus erythematosus sometimes occurs, one of the reasons why Ackerman regards this condition as a variant of lupus erythematosus. Two cases of mycosis fungoides that clinically mimicked REM have been reported. 228
One study has shown that fibroblasts from lesional skin exhibit an abnormal response to exogenous interleukin-1β, 229 but a more recent study has shown that the accumulation of hyaluronan in REM may be related to populations of factor XIIIa+/HAS2+ dermal dendrocytes rather than to dermal fibroblasts. 230 HAS2 is one of three genetically distinct isoforms of hyaluronan synthase (HAS), the enzyme thought to be responsible for the synthesis of hyaluronan, a major component of the dermal matrix. 230
Topical tacrolimus and UVA1 radiation have been found to be a safe alternative to systemic antimalarials in the treatment of this condition.229. and 230.
There is a mild superficial and mid-dermal perivascular infiltrate with variable deep perivascular extension, the latter sometimes being restricted to the region of the eccrine glands (Fig. 13.5). 220 There may be some perifollicular infiltrate as well. This infiltrate is predominantly lymphocytic (helper T cells), 233 with a few admixed mast cells, histiocytes, and factor XIIIa-positive dendrocytes. 232 There is slight vascular dilatation and, sometimes, focal mild hemorrhage in the upper dermis. 225
Reticular erythematous mucinosis. (A) There is a superficial and deep infiltrate of lymphocytes and an increase in interstitial mucin. (B) This further example shows overlap features with tumid lupus erythematosus although clinically the patient initially presented as reticular erythematous mucinosis. (H & E)
There is separation of dermal collagen bundles, and variable amounts of stringy basophilic mucin can be seen predominantly in the upper and mid dermis (Fig. 13.6). The mucin is most conspicuous around the infiltrate and appendages and within the upper dermis. 224 A few stellate cells may be present. The epidermis is usually normal, although mild exocytosis with spongiosis and focal lichenoid inflammation have been reported. 224
Reticular erythematous mucinosis. There is increased mucin throughout the dermis. (Alcian blue)
The mucin gives variable staining reactions. Colloidal iron staining is superior to Alcian blue, which on occasions has failed to demonstrate mucin.234. and 235. The material is not usually metachromatic with toluidine blue. 235 Staining with colloidal iron and Alcian blue will be negative following digestion with hyaluronidase. There may be focal fragmentation of elastic fibers. 234 The mucin is largely hyaluronan, which is not well fixed by formalin. 232
Direct immunofluorescence has shown the deposition of immunoglobulins, particularly IgM, and some C3, along the basal layer in several cases,236.237. and 238. lending support for the view that REM is a subset of lupus erythematosus.
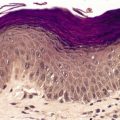
Scleredema (scleredema adultorum of Buschke) is characterized by the development of non-pitting induration of the skin with a predilection for symmetrical involvement of the posterior neck, the shoulders, the upper trunk, and the face.241. and 242. Localization to the thighs and periorbital region have been reported.243. and 244. In cases of more widespread involvement, the upper part of the body is always involved much more than the lower part, and the feet are spared. The condition occurs at all ages, although nearly 50% of cases develop in children and adolescents. 245 There is a predilection for females.
There are several different clinical settings.246. and 247. In one group the onset is sudden and follows days to weeks after an acute febrile illness caused by streptococci or viruses. A case occurring in association with HIV infection has been reported. 11 It has followed scabies infestation complicated by secondary bacterial infection with streptococci. 248 Spontaneous resolution occurs in approximately one-third of the post-infectious group after 6–18 months. This group is now seen less frequently than previously. Another clinical group has an insidious onset, without any predisposing illness, and a protracted course. The third group is associated with insulin-dependent, maturity onset diabetes, which is difficult to control.249.250.251. and 252. Vascular complications of diabetes are common. 253 It can also be associated with type 2 diabetes.254. and 255. A fourth group is associated with a monoclonal gammopathy.256.257.258.259.260.261.262.263.264.265. and 266. Extracorporeal photopheresis and chemotherapy have been used to treat such cases.267. and 268. Other associations are mechanical stress, 269 carcinoma of the gallbladder, 270 and rheumatoid arthritis. 271 Scleredema is insidious in onset and prolonged in its course.
Systemic manifestations such as ECG changes, serosal effusions, and involvement of skeletal, ocular and tongue musculature may develop. Cutaneous abscesses or cellulitis may precede or follow the onset of the condition.272. and 273. Rarely, there is erythema at sites of skin thickening. 274 Two cases with overlap features between scleredema and stiff-skin syndrome have been reported. 275
Scleredema is characterized by the accumulation of glycosaminoglycans, particularly hyaluronan, in the dermis with concurrent dermal sclerosis. The etiology is unknown, although fibroblasts from the fibrotic skin of patients with scleredema show enhanced collagen production and elevated type I procollagen messenger RNA levels in the cultured fibroblasts. 258 This suggests that fibroblasts from involved skin have a biosynthetically activated phenotype. 276
Scleredema has been successfully treated with UVA1 phototherapy. 277
The epidermis is usually unaffected except for some effacement of the rete ridge pattern and occasionally mild basal hyperpigmentation. 278 Hyperkeratosis is rare. 279 There is thickening of the reticular dermis, with collagen extending also into the subcutis. The collagen fibers are swollen and separated from one another. The extent of this separation, which mirrors the amount of interstitial mucopolysaccharide present, depends on the stage of the disease (Fig. 13.7). This material may only be present in noticeable amounts at the onset of the disease. 247 Multiple biopsies and the use of several stains may be necessary to demonstrate the dermal mucin. 280 Sometimes it is most prominent in the lower dermis. 281 Cetylpyridinium has been proposed as a superior fixative to formalin for the preservation of the interstitial mucopolysaccharides. 245 These may subsequently be stained with Alcian blue or toluidine blue (pH 5.0 or 7.0) or with colloidal iron. Cryostat sections of unfixed material usually result in the optimal preservation of the interstitial hyaluronan.
Scleredema. (A), (B) The collagen bundles are slightly swollen and separated from one another. Cellularity of the dermis is normal with no increase in fibroblasts. (H & E) (C), (D) Mucin deposits are present on the surface of the collagen. (Colloidal iron)
Other features of scleredema include preservation of the appendages and an increase in mast cells. 282 Other inflammatory cells are sparse. Elastic fibers are reduced and may be fragmented. 278
The acellular fibrosis of the dermis in scleredema contrasts with the marked fibroblastic proliferation seen in scleromyxedema and the patchy deep inflammatory cell infiltrate seen at the advancing edge of plaques of morphea.
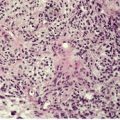
Focal mucinosis usually presents as a solitary, asymptomatic, flesh-colored papule or nodule on the face, trunk, or proximal and mid extremities of adults.283.284.285. and 286. The nodules average 1 cm in diameter. There has been a report of multiple nodules localized to a ‘palm-wide area’ of the right leg287 and also a report of multiple lesions in a patient with hypothyroidism, which responded to thyroxine. 94 Digital lesions with the histology of focal mucinosis have been described in scleroderma (see p. 310). It has been reported in association with other cutaneous mucinoses. 288 Oral focal mucinosis is a related condition. 289 The case reported as ‘reactive pseudotumoral papular mucinosis’ is best regarded as a variant of focal mucinosis. 290
It is thought that increased amounts of hyaluronan are produced by fibroblasts at the expense of the connective tissue elements. 291
There is a slightly elevated or dome-shaped dermal nodule with separation and variable replacement of collagen bundles by mucinous deposits (Fig. 13.8). These deposits may be localized to the upper dermis or extend through the full thickness of the dermis. The subcutis is rarely involved. 292 Slit-like spaces occasionally develop. The margins of the mucinous deposition are not sharply demarcated. Spindle-shaped fibroblasts are present within the mucinous areas and there may be an increase in small blood vessels. The appearances resemble the early stages of a digital mucous cyst (see below) and an individual lesion of papular mucinosis. The material stains with colloidal iron and Alcian blue at pH 2.5 and is metachromatic with toluidine blue at pH 3.0 (Fig. 13.9). Immunohistochemistry confirms that the spindle cells are predominantly fibroblasts with some factor XIIIa-positive dendritic cells. 285
Focal mucinosis. There is a large ‘pool’ of mucin dispersed through much of the dermis. This is the nodular variant. (H & E)
Focal mucinosis. The abundant mucin is confirmed with a mucin stain. (Colloidal iron)
The fibroblasts have a well-developed rough endoplasmic reticulum. In addition, there are large macrophages and granular and amorphous material representing the mucinous deposits. 287
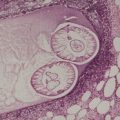
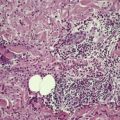
Digital mucous cysts occur as solitary, dome-shaped, shiny, tense cystic nodules on the dorsum of the fingers, usually involving the base of the nail. 293 Subungual cases, distal to the nailfold, also occur. 294 Multiple cysts on a single finger have been reported; they were of the ganglionic subtype (see below). 295 The toes are uncommonly involved. The cysts are found in the middle-aged or elderly and there is a slight female preponderance. A second type, overlying the distal interphalangeal joint, is related to a ganglion as injection studies have demonstrated a connection with the underlying joint cavity. 296 It has been suggested that all digital mucous cysts connect with this joint. 297 Ganglions at other sites are beyond the scope of this book.298. and 299. A superficial angiomyxoma arising on the finger has been reported; it mimicked a digital mucous cyst. 300
A skin flap that includes the undersurface of the cyst and the tissues between the cyst and the distal interphalangeal joint, but avoiding removal of the skin itself, has been used successfully in their treatment. 297 Infrared coagulation has also been used. 301
The variant developing at the base of the nail resembles focal mucinosis with a large myxoid area containing stellate fibroblasts, sometimes with microcystic spaces (Fig. 13.10). The overlying epidermis may be thinned by the expanding subepidermal collection of mucus. The mucin stains with the colloidal iron stain, as well as with Alcian blue at pH 2.5, and it is digested by hyaluronidase. 302
Digital mucous cyst. There is a ‘pool’ of mucin containing many fibroblasts. This variant resembles focal mucinosis. (H & E)
The ganglionic variant comprises a cystic space with a well-defined fibrous wall of variable thickness and density (Fig. 13.11). There are often small areas of myxoid change adjacent to the wall. 303 There may be an attenuated synovial lining.
Digital mucous cyst. This is the ganglionic variant. (H & E)
Mucoceles (mucous cysts) result from the rupture of a duct of a minor salivary gland with extravasation of mucus into the submucosal tissues, most commonly of the lower lip. 304 They may also develop in the buccal mucosa or tongue. Multiple superficial mucoceles on the lips and oral mucosa have been reported in association with graft-versus-host disease. 305 A case reported on the anterior neck was related to the presence of ectopic minor salivary glands. 306 Mucoceles are found mostly in young adults.
Mucoceles are translucent, whitish or bluish nodules with a firm cystic consistency and vary in size up to 1 cm in diameter. They occasionally rupture spontaneously or after minor trauma.
A superficial variant of mucocele, which results in vesicular lesions that may be mistaken for mucous membrane pemphigoid, has been reported. 307 They may be single or multiple and they arise on non-inflamed mucosa.
Two patterns may be seen, but there may be some overlap between them. 304 In one, there is a cystic space with a surrounding poorly defined lining of macrophages, fibroblasts, and capillaries with variable amounts of connective tissue. In the other pattern, there is granulation and fibrous tissue containing mucin-filled spaces with variable numbers of muciphages (Fig. 13.12). Small cystic spaces may be present. Numerous neutrophils and some eosinophils are present in the cystic spaces or stroma of both types. There is no epithelial lining to the cyst, although occasionally a ruptured salivary duct may be seen at one edge of the cyst. Minor salivary gland tissue is present in the adjacent connective tissue.
Mucocele. (A) A duct leads into a pseudocyst lined by granulation tissue and muciphages. (B) The lumen contains mucus admixed with inflammatory cells. (H & E)
The mucin is strongly PAS positive and diastase resistant and is positive with Alcian blue at pH 2.5 and with colloidal iron.
Superficial mucoceles are subepithelial although there may be partial or complete epithelial regeneration across the vesicle floor. They contain sialomucin. Salivary gland ducts are present in the immediate vicinity of the lesions and are a clue to the diagnosis. 307
Cutaneous myxomas have been reported in approximately 50% of the patients with the complex of cardiac myxomas, spotty pigmentation (lentigines and blue nevi), and endocrine overactivity.308.309.310.311.312. and 313. This combination of lesions is known as Carney’s complex (OMIM 160980), an autosomal dominant condition which has been mapped via linkage analysis to chromosome 2p16 and 17q2. 314 A mutation in the PRKAR1α gene on the chromosome 17q22–q24 locus has been identified in patients with Carney’s complex, particularly those with atrial myxomas. This gene, which is a tumor-suppressor gene, encodes the R1-α regulatory subunit of cyclic adenosine monophosphate-dependent protein kinase A (PKA). 315 The gene located on 2p16 has not yet been identified.
The cutaneous myxomas may be the earliest manifestation of the syndrome. 316 They are typically found on the eyelids, nipples and buttocks, and in the external ear canals; they are frequently misdiagnosed as fibroepithelial lesions. 317 Spotty pigmented lesions (lentigines and blue nevi) are typically found on the face, specifically along the lips and around the eyes.317. and 318. Similar cases have been reported as the NAME or LAMB syndrome (see Ch. 32, p. 710).
Solitary and disseminated myxomas unassociated with any systemic abnormalities may also occur.319.320.321.322. and 323. They are benign neoplasms, but they are included here because of their prominent stromal mucin. 324 They are probably the same entity as solitary superficial angiomyxoma (see Ch. 34, p. 844).
In one reported case, a patient with multiple periorbital myxomas progressed to a scleromyxedema-like dermatosis. 325
The tumors are sharply circumscribed, non-encapsulated lesions which may be in the dermis or subcutis. They are composed of a prominent mucinous matrix containing variably shaped fibroblasts, prominent capillaries, mast cells, and a few collagen and reticulin fibers (Fig. 13.13). Sometimes an epithelial component is present and this may take the form of a keratinous cyst or epithelial strands with trichoblastic features. 310These latter changes may mimic the basaloid proliferations sometimes seen above dermatofibromas. 326 The lesions differ from focal mucinosis by their vascularity. If the vascular component is marked, the term ‘angiomyxoma’ is often used (see p. 844). 321 Nerve sheath myxomas are more cellular, often with a distinct patterned arrangement.
Myxoma. There are thin collagen bundles and fibroblasts scattered through a mucinous matrix. (H & E)
The designation ‘fibromyxoma’ has been applied to the lesions in a patient with multiple cutaneous tumors, resembling dermatofibromas clinically but containing more fibroblasts than the usual cutaneous myxomas and some histiocytes, in addition to the interstitial mucin. 327 Familial myxovascular fibroma is a morphologically related entity (see p. 813).
Nevus mucinosus (mucinous nevus) is a variant of connective tissue nevus in which there is a deposition of acid mucopolysaccharides (proteoglycans) in the dermis.142. and 328. The existence of such an entity has been postulated for some time in the light of the nevoid lesions of the other connective tissue elements – collagen and elastic tissue. Cases previously reported as cutaneous mucinosis of infancy (see above) may represent examples of nevus mucinosus.
The lesions are small papules, in a grouped, zosteriform or linear arrangement, on the extremities or trunk.329.330. and 331. They are present at birth or appear in childhood or early adult life. 332 A familial case has been reported. 333
The epidermis usually shows some acanthosis with thin, elongated rete ridges. There is a grossly thickened papillary dermis with an ‘empty appearance’ due to the deposition of abundant acid mucopolysaccharides. The mucinous stroma is strongly stained with hyaluronate-binding protein, indicating the presence of hyaluronate. 334 Fibroblasts are slightly increased in number, with occasional stellate forms. 142 Elastic fibers are decreased in the papillary dermis but normal in the deeper dermis. 331 A sparse inflammatory infiltrate composed of lymphocytes and eosinophils may be present in the mid dermis beneath the mucin deposit. 334
The mucin in nevus mucinosus is deposited in the upper dermis, in contrast to papular mucinosis where the mucin is at a lower level. Nevus mucinosus differs from acral persistent papular mucinosis on clinical grounds and also by having fewer fibroblasts within the lesions. Focal mucinosis differs by its larger size and solitary nature.
Progressive mucinous histiocytosis (OMIM 142630), a rare, autosomal dominant histiocytosis of childhood, is characterized by multiple small papules composed of epithelioid and spindle-shaped histiocytes set in abundant stromal mucin. Sporadic cases have been reported. 335 This entity is discussed in more detail with the histiocytoses (see p. 956).
Mucin deposition may be present in a wide variety of connective tissue diseases and in some tumors. These conditions include dermatomyositis, lupus erythematosus (see below), scleroderma, 336 linear morphea, 337 the toxic oil syndrome, hypertrophic scars, connective tissue nevi, granuloma annulare, malignant atrophic papulosis (Degos’ disease), erythema annulare centrifugum, postinflammatory hyperpigmentation, 338 Still’s disease, 339 mycosis fungoides, 340 and Jessner’s lymphocytic infiltrate.4. and 5. Localized mucinosis may follow erysipelas, 341 or be associated with lower leg edema and morbid obesity. 31 Mucin is sometimes present in large amounts around the secretory coils of the eccrine glands of the lower leg when sweat excretion is blocked. 5 Tumors containing mucin include neurofibromas, neurilemmomas, nerve sheath myxomas, chondroid syringomas, and some basal cell carcinomas. A diffuse dermal mucinosis can be found occasionally in biopsies of lesional skin from patients with discoid and systemic lupus erythematosus (SLE).342. and 343. Rarely, patients with SLE and systemic sclerosis can present with papulonodules or plaques due to a diffuse mucinous deposition in the skin (Fig. 13.14).336.343.344.345.346.347.348.349.350. and 351. In these individuals, the deposition occurs in areas free from specific lesions of lupus erythematosus and it produces clinically distinct manifestations. A factor (or factors) in the patient’s serum appear(s) to stimulate fibroblasts to produce increased amounts of glycosaminoglycan. 347 Multiple plaques of mucinosis have been reported as the sole manifestation of dermatomyositis in a patient with a nasopharyngeal carcinoma. 352 A solitary plaque on the face has been reported in a patient with secondary extramedullary cutaneous plasmacytoma. 353
(A) Lupus mucinosis. (H & E) (B) There is wide separation of attenuated collagen bundles by mucin. (Colloidal iron) (Photographs kindly provided by Dr Geoffrey Strutton)
The case reported as ‘plaque-like erythema with milia’ occurred in a renal transplant recipient on cyclosporine (ciclosporin), which was implicated in the pathogenesis. There were extensive mucin deposits in the dermis. 354
Finally, patients with mitral valve prolapse in which myxomatous degeneration of mitral valve leaflets occurs, have been found to have increased cutaneous deposits of proteoglycan (mucin) in the skin. 355
Follicular mucinosis is a tissue reaction pattern in which hair follicles and the attached sebaceous gland accumulate mucin with some dissolution of cellular attachments (Fig. 13.15). 3 There is an accompanying perifollicular and perivascular inflammatory cell infiltrate of lymphocytes, histiocytes, and a few eosinophils. Sometimes follicles are converted into cystic cavities with disruption of much of the external root sheath. These cysts contain mucin, inflammatory cells, and keratinous debris. There is often a marked disparity between the amount of follicular mucin and the degree of follicular and perifollicular inflammation. 356 The material is stained by the Alcian blue and colloidal iron methods. Follicular mucinosis is sometimes seen in arthropod bite reactions and in the exaggerated bite reactions that occur in some patients with chronic lymphocytic leukemia357 (see secondary follicular mucinosis below).
Follicular mucinosis. The accumulation of mucin within the hair follicle has resulted in the dissolution of many cellular attachments. (H & E)
The concept that follicular mucinosis is a tissue reaction pattern and not a disease sui generis is a relatively recent one3 and, as a consequence, most reports in the literature use the term ‘follicular mucinosis’ for what Pinkus described as alopecia mucinosa in 1957. 358
Follicular mucinosis (alopecia mucinosa) is an uncommon inflammatory dermatosis with a predilection for adults in the third and fourth decades of life. 359 A congenital case has been reported. 360 Three clinical types have traditionally been recognized:361. and 362. a benign transient form with one or several plaques or grouped follicular papules, usually limited to the face or scalp and with accompanying alopecia;363.364.365.366.367. and 368. a more widely distributed form with follicular papules, plaques, and nodules on the extremities, face, and trunk and a course often exceeding 2 years; 369 and a third group, accounting for 15–30% of cases, with widespread lesions and associated with malignant lymphoma of the skin or mycosis fungoides.370.371.372.373. and 374. Rarely, patients with leukemia cutis, 375 leukemia without skin lesions, 376 Hodgkin’s disease, 377 familial reticuloendotheliosis, 378 Sézary syndrome,379. and 380. squamous cell carcinoma of the tongue, 381 and angiolymphoid hyperplasia382. and 383. have also had this presentation. This third group was regarded by Hempstead and Ackerman3 as belonging to the group of secondary follicular mucinoses because of the lack, at that time, of convincing examples of follicular mucinosis (alopecia mucinosa) progressing to mycosis fungoides or cutaneous lymphoma.384. and 385. However, cases of follicular mucinosis progressing to mycosis fungoides have since been well documented.386. and 387. A study of T-cell clonality in follicular mucinosis has shown a monoclonal T-cell population in all patients with associated cutaneous T-cell lymphoma and also in 9 of 16 patients with the primary form of the disease. 388 In another series of 4 cases, presenting as an acneiform eruption of the face in early adulthood, 2 cases have demonstrated a clonal rearrangement of the T-cell receptor within the cutaneous infiltrate. 389 A more recent study from Graz, Austria, has also cast doubt on the validity of the continued separation of the so-called ‘primary form’ of follicular mucinosis from the lymphoma-associated group. Not only was there some clinical overlap between the two groups, but histopathological examination did not allow differentiation of the two groups.390. and 391. Furthermore, a monoclonal rearrangement of the TCR gene was demonstrated by PCR analysis in 4 of 10 cases from the primary group and 7 of 17 cases from the lymphoma-associated group. 390 These authors postulate that even the primary form of follicular mucinosis may be a form of localized cutaneous T-cell lymphoma.390. and 392. These cases raise the question – when does follicular mucinosis become mycosis fungoides?393. and 394. Ackerman and colleagues have also proposed that alopecia mucinosa is mycosis fungoides.395. and 396. Contrary views still exist. 397 LeBoit has challenged the view that there are only two choices – inflammatory disease or mycosis fungoides – for the pathogenesis of follicular mucinosis (alopecia mucinosa). He has raised the possibility that its lesions are ‘an expression of a self-limited proliferation of lymphocytes, e.g. a benign neoplasm of them’. 398 He also points out that lymphocytic infiltrates are the only ones in which clonality has been used to infer malignancy. 398 Guitart and Magro have included idiopathic follicular mucinosis in their classification of cutaneous T-cell lymphoid dyscrasias, a unifying term for idiopathic chronic dermatoses with persistent T-cell clones. 399 This is another ‘third choice’, and the one favored by the author of this book.
The protean clinical presentations attributed to follicular mucinosis, such as eczematous, 400 annular, pityriasis rosea-like, 401 and folliculitis, are in many cases examples of specific dermatoses in which secondary follicular mucinosis is present.
Although it has been postulated that follicular keratinocytes are the source of the mucopolysaccharides,3. and 402. this has not been confirmed in another study, which proposed a role for cell-mediated immune mechanisms in the etiology. 385 A more recent study has found active secretion of hyaluronate by follicular keratinocytes in follicular mucinosis, but CD44 expression (a cell-surface receptor of hyaluronate) was not increased. 403
Primary follicular mucinosis has been successfully treated with photodynamic therapy. 404
The major changes are those described above for the tissue reaction known as follicular mucinosis (Fig. 13.16). However, the inflammatory cell infiltrate is predominantly follicular, perifollicular, and perivascular in location, in contrast to the follicular mucinosis secondary to lymphomas where the infiltrate is more dispersed, often heavier and nodular, and sometimes has more plasma cells and fewer eosinophils than in primary follicular mucinosis. 356 Even the follicular mucinosis of mycosis fungoides can have an unimpressive infiltrate, composed of small lymphocytes with minimal atypia. 407 Furthermore, there is usually a milder mucinous change in follicular mucinosis related to lymphomas than in primary follicular mucinosis. However, as mentioned above, the study from Graz casts doubt on the validity of separating the primary form from the lymphoma-associated group. 390 Atypical cells and Pautrier microabscesses are not seen in follicular mucinosis, 356 but may be present in secondary follicular mucinosis with accompanying lymphoma or mycosis fungoides. Rarely, dermal mucinosis377. and 408. or a proliferation of eccrine sweat duct epithelium is present. 409 The term folliculotropic T-cell lymphocytosis has been proposed for a case that clinically resembled follicular mucinosis, but in which there were minimal mucin deposits in the hair follicles. 410
Follicular mucinosis (alopecia mucinosa). Multiple follicles show the reaction pattern of follicular mucinosis. (H & E)
Follicular mucinosis may be found as an incidental phenomenon in rare cases of lichen simplex chronicus, hypertrophic lichen planus, discoid lupus erythematosus, acne vulgaris, pseudolymphoma, nevocellular nevi, 412 and arthropod bite reactions. 3 Cases associated with the ingestion of various drugs, such as calcium channel blockers, beta-blockers, antihistamines, antidepressants, and imatinib, have been reported. 413 The superficial follicular spongiosis seen in some cases of atopic dermatitis, Grover’s disease, and actinic prurigo, 414 as well as in infundibulofolliculitis, may contain small amounts of mucin. 415
There is one report of an adolescent male who developed two plaques on the face characterized by prominent perifollicular mucin. The lesions were regarded as being of nevoid origin. 416 Perifollicular mucinosis has also been reported in a patient with HIV infection and an atypical pityriasis rubra pilaris-like eruption. 417
Small foci of intercellular mucin are an inconstant and incidental finding in some spongiotic dermatoses, as well as in verrucae, seborrheic keratoses, basal cell and squamous cell carcinomas, and keratoacanthomas. 3 Epidermal mucin is sometimes a conspicuous feature in mycosis fungoides (see p. 977).
Eccrine ductal mucinosis, in which there was mucin between the cells of the outer layer of the eccrine duct, has been reported in a patient with HIV infection and probable scabies. The significance of these related conditions and the nature of the mucinosis remain an enigma. 418
The mucopolysaccharidoses are a group of 10 lysosomal storage diseases which result from the deficiency of specific lysosomal enzymes involved in the degradation of dermatan sulfate, heparan sulfate, or keratan sulfate, singly or in combination.419.420. and 421. As a consequence, mucopolysaccharides accumulate in various tissues and are excreted in the urine. 422 Because of genetic variability, heterogeneity, and pleiotropism, more than 10 clinical syndromes are associated with the 10 enzyme deficiencies. 419
The specific enzyme defect can be identified using cultured fibroblasts, although in many instances a tentative diagnosis is possible on the clinical features alone. Prenatal diagnosis is possible on both cultured amniotic fluid cells and chorionic villus samples. Analysis of the urine for certain mucopolysaccharides will also assist in the diagnosis. The urine contains heparan sulfate in the Sanfilippo syndrome (MPS III – OMIM 252900), keratan sulfate in the Morquio syndrome (MPS IV – OMIM 253000), dermatan sulfate in the Maroteaux–Lamy syndrome (MPS VI – OMIM 253200), and an excess of dermatan and heparan sulfates in varying ratios in the others.
The mucopolysaccharidoses share many clinical features. 419 These include skeletal abnormalities characterized as dysostosis multiplex (except in Morquio’s syndrome), short stature (excluding Scheie’s syndrome – MPS IS – OMIM 607016), corneal clouding (except in the Hunter (MPS II) and Sanfilippo (MPS III) syndromes), deafness, grotesque facies (gargoylism), hirsutism, premature arteriosclerosis, hepatosplenomegaly, and severe mental retardation (excluding MPS VI and MPS IS). 419 The best known of the mucopolysaccharidoses are Hurler’s syndrome (MPS I – OMIM 607014), which has the worst prognosis and is due to a deficiency of α-L-iduronidase encoded by the IDUA gene at 4p16.3, 423 and Hunter’s syndrome (MPS II – OMIM 309900), which results from a deficiency of iduronate-2-sulfate sulfatase. Hunter’s syndrome is the only X-linked mucopolysaccharidosis with the gene defect located at Xq28.424. and 425. The genetic defect in Sanfilippo syndrome type A (OMIM 252900) is situated on chromosome 17q25.3. Numerous different mutations have been identified. 426 Recently, the transmembrane protein 76 gene (TMEM76) has been identified as a possible cause of Sanfilippo syndrome type C (OMIM 252930), a disorder of heparan sulfate degradation due to a deficiency of the enzyme N-acetyltransferase. 427 Other publications attribute this variant to mutations in the HGSNAT gene.
Cutaneous manifestations of the mucopolysaccharidoses include hirsutism and dryness of the skin. There may be mild thickening also; this is most marked in Hurler’s syndrome where sclerodermoid thickening of the fingers and furrowing of the skin can occur.428. and 429. Firm, flesh-colored to waxy papules and nodules can be found on the upper trunk, particularly in the scapular region, in Hunter’s syndrome.424.430.431.432. and 433. They give rise to a characteristic cobblestone appearance. 434 These papules disappear after a hemopoietic stem cell transplant. 435 Extensive Mongolian spots are another manifestation of Hunter’s syndrome.436. and 437.
Allogeneic bone marrow transplantation before the age of 2 years will halt disease progression in Hurler’s syndrome. A recombinant human alpha-L-iduronidase has been given intravenously to patients with this deficiency.
Metachromatic granules are present in the cytoplasm of fibroblasts in all cases and are sometimes seen in eccrine sweat glands and epidermal keratinocytes. 428 Metachromatic granules were absent in one recently reported case, but the large amount of extracellular mucin may have obscured their recognition. 434 Extracellular mucin of any significant amount is only found in the mid and lower dermis in the papulonodules of Hunter’s syndrome. 430 There are widely separated collagen bundles in the dermis with intervening pale blue, fibrillar material. 425 This mucin can be seen in toluidine blue-stained sections of alcohol-fixed material, but it can also be demonstrated with the Giemsa, Alcian blue, and colloidal iron stains.425. and 430. The fibroblasts are slightly more prominent than usual with an oval nucleus and a definable cytoplasmic outline, but these changes are quite subtle.