Key Words
internal disease, necrobiosis lipoidica, granuloma annulare, acanthosis nigricans, xanthomas, dyslipoproteinemia, neurofibromatosis, tuberous sclerosis, multiple hamartoma syndrome, renal carcinoma, diabetes mellitus, erythematous plaques, xanthelasma, plane xanthomas, hyperlipoproteinemia
Certain cutaneous diseases are frequently associated with internal disease. The skin disease itself may be inconsequential, but its presence should prompt investigation of possible related internal disorders. A selected group of such diseases is discussed in this chapter. Pigmentary skin changes associated with internal diseases are also discussed in Chapter 19 .
Cutaneous Manifestations of Diabetes Mellitus
Approximately 30% of patients with diabetes mellitus develop a skin disorder sometime during the course of disease. A list of these disorders follows:
- •
Candida infections (mouth, genital)
- •
Carotenodermia (yellow skin)
- •
Diabetic bullae
- •
Diabetic dermopathy (shin spots)
- •
Diabetic thick skin
- •
Erythema (face, lower legs, feet)
- •
External otitis
- •
Finger pebbles
- •
Foot ulcers
- •
Acanthosis nigricans (insulin resistance syndromes)
- •
Gas gangrene (nonclostridial)
- •
Granuloma annulare (localized or generalized)
- •
Insulin lipodystrophy
- •
Necrobiosis lipoidica
- •
Yellow nails
- •
Perforating disorders
- •
Eruptive xanthomas
Necrobiosis Lipoidica
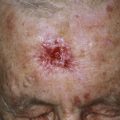
Necrobiosis lipoidica (NL) is a disease of unknown origin, but more than 50% of the patients with NL are generally insulin dependent. The average age at onset is 30 years. It is frequently associated with diabetes. More than 75% of patients with NL either have or will develop diabetes mellitus. However, it is seen in only 0.3% to 0.7% of the entire diabetic population. The skin lesions may appear years before the onset of diabetes, and most patients with diabetes do not develop NL. The disease may occur at any age, but it most commonly appears in the third and fourth decades. Most of the patients are females, and in most cases the lesions are confined to the anterior surfaces of the lower legs ( Fig. 26.1 ).
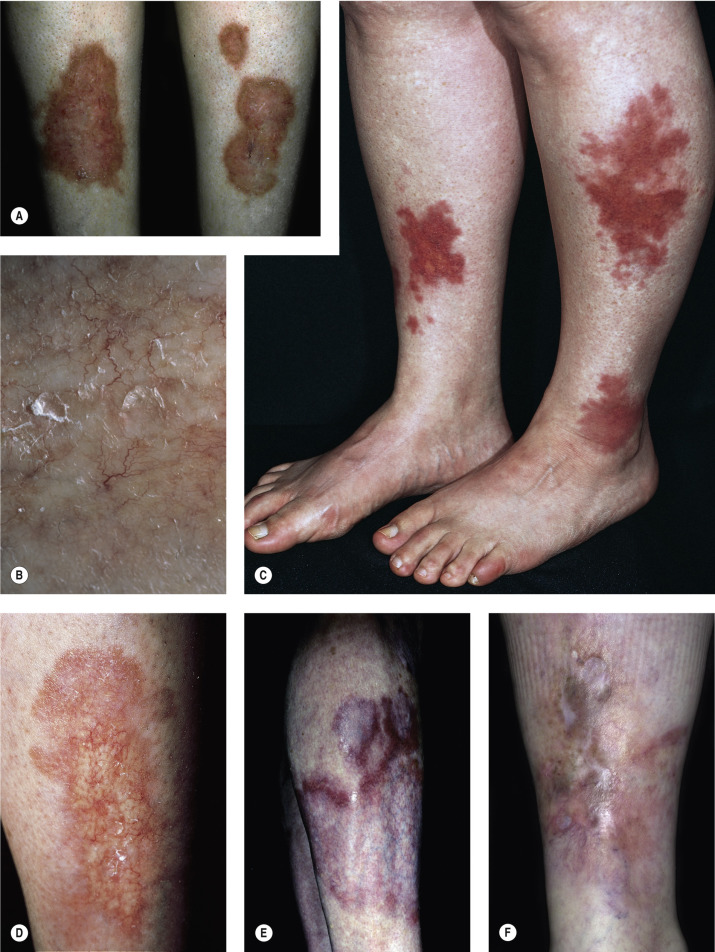
The eruption begins as an oval, violaceous patch and expands slowly. The advancing border is red, and the central area turns yellow-brown. The central area atrophies and has a waxy surface; telangiectasias become prominent (see Fig. 26.1 ). Ulceration occurs, particularly following trauma, in 13% of cases. In many instances the clinical presentation is so unique that biopsy is not required. Squamous cell carcinoma may arise within NL diabeticorum that has been present for years or is chronically ulcerated.
Treatment.
Currently, there are no evidence-based guidelines for the treatment of NL. A complete review of all treatments is found in reference .
Topical and Intralesional Steroids.
Topical and intralesional steroids arrest inflammation but promote further atrophy. Clobetasol propionate under occlusion has successfully treated thick plaques. Intralesional injections effectively control small areas of NL, but the concentration of triamcinolone acetonide (10 mg/mL) should be diluted with saline or Xylocaine to 2.5 mg/mL to avoid atrophy.
Systemic Corticosteroids.
A 3- to 5-week course of systemic corticosteroids may arrest the disease; however, restitution of atrophic skin lesions is not achieved. Ulceration of NL can at times be successfully treated with oral prednisolone.
Pentoxifylline.
Pentoxifylline 400 mg three times a day is reported to result in significant improvement after 1 month of treatment. Ulcerating NL is reported to respond within 8 weeks of administration of 400 mg of pentoxifylline twice a day. Pentoxifylline is thought to decrease blood viscosity by increasing fibrinolysis and red blood cell deformability and also to inhibit platelet aggregation.
Aspirin and Dipyridamole.
Low-dose aspirin and dipyridamole are thought to inhibit platelet aggregation, but reports concerning their efficacy in healing ulcers in plaques of NL are conflicting. The recommended treatment is aspirin 3.5 mg/kg every 48 hours, which for the average patient is 325 g daily (one tablet); or dipyridamole (25-, 50-, or 75-mg tablets) 2 to 3 mg/kg/day, which for the average patient is 150 to 200 mg daily in divided doses. For effective control of ulceration, platelet inhibition therapy must be used for a minimum of 3 to 7 months. Recommended treatment schedules should be followed because there is evidence that higher dosages can decrease treatment effectiveness.
Other Treatments.
Cyclosporine, tacrolimus ointment 0.1%, etanercept, mycophenolate mofetil, fumaric acid esters, photochemotherapy with topical psoralen plus ultraviolet A (PUVA), chloroquine (200 mg/day), hydroxychloroquine (400 mg/day), infliximab, and thalidomide (150 mg/day) are reported to be effective.
Skin Grafting.
Skin grafting is effective for extensive disease.
Granuloma Annulare
There are conflicting reports about the association of granuloma annulare (GA) with diabetes mellitus. Most patients with the localized form of GA do not have clinical or laboratory evidence of diabetes. The association between disseminated GA and diabetes has been established, but the frequency is unknown. In a retrospective study 12% of patients with GA had diabetes mellitus. Those patients suffered significantly more often from chronic relapsing GA than nondiabetic patients. Granuloma annulare can be associated with human immunodeficiency virus (HIV) and can present at all stages of HIV infection. Generalized GA is the most common clinical pattern in HIV infection.
Clinical Presentation.
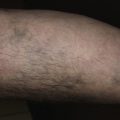
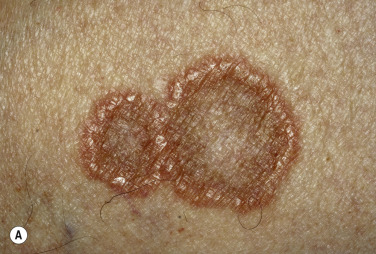
Granuloma annulare is characterized by a ring of small, firm, skin-colored or red papules. The localized form, most common in young adult females, is most frequently found on the lateral or dorsal surfaces of the hands and feet ( Fig. 26.2 ). The disease begins with an asymptomatic, skin-colored papule that undergoes central involution. Over months, a ring of papules slowly increases in diameter to 0.5 to 5 cm. The duration of the disease is highly variable. Many lesions undergo spontaneous involution without scarring, whereas others last for years. The familial occurrence of GA is uncommon but has been noted in siblings, twins, and successive generations.
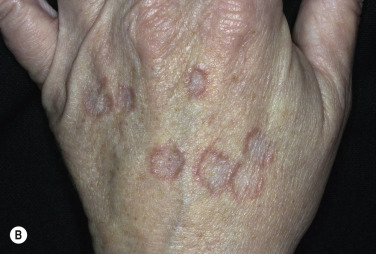
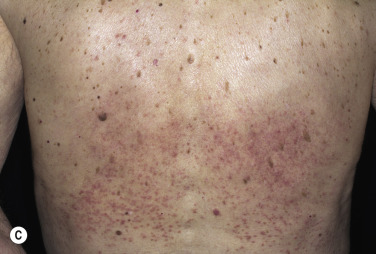
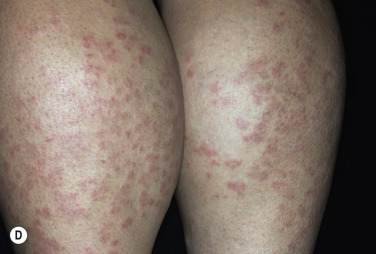
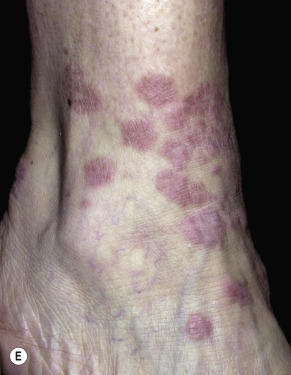
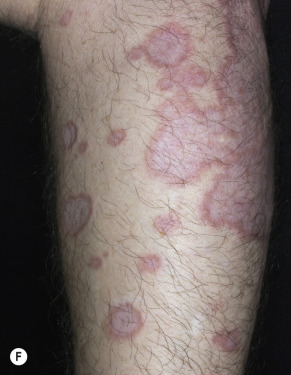
Disseminated GA occurs in adults and appears with numerous skin-colored or erythematous papules, some of which form annular rings. The papules may be accentuated in sun-exposed areas. The course is variable; many lesions persist for years.
Generalized perforating GA is characterized by 1- to 4-mm umbilicated papules on the extremities and is most commonly seen in children and young adults. Biopsy shows transepithelial elimination of degenerating collagen fibers. GA occurred during anti–tumor necrosis factor (anti-TNF) therapy (4.5% of 119 patients) (infliximab, adalimumab, etanercept) for rheumatoid arthritis. Patients who develop GA usually heal, remain remarkably healthy, and do not ordinarily develop other odd diseases.
Subcutaneous GA occurs in children. Painless subcutaneous nodules in the lower anterior tibial region or foot and in the scalp, typically in the occiput, are the most common presenting features. The mean age at presentation is 3.9 years. Diagnosis requires an excisional biopsy. Lesions may resolve spontaneously and recur after excision. No record of progression to systemic illness is reported.
Diagnosis.
The clinical presentation is characteristic, and biopsy may not be required. Histologic examination shows collagen degeneration, a feature similar to that seen in NL.
Treatment.
Localized lesions are asymptomatic and are best left untreated. Those patients troubled by appearance may be treated with intralesional injections of triamcinolone acetonide (2.5 to 5 mg/mL). The solution should be injected only into the elevated border. Topical steroids have little effect. Localized lesions have responded to imiquimod cream. Disseminated GA has been reported to respond to dapsone, isotretinoin, acitretin, hydroxychloroquine, niacinamide (1.5 g/day), fumaric acid esters, hydroxyurea, narrowband ultraviolet B therapy, and psoralen and PUVA therapy. Four patients with disseminated GA were treated with a cycle of cyclosporine therapy for 6 weeks. Cyclosporine was started at a dose of 4 mg/kg/day for 4 weeks, and subsequently reduced by 0.5 mg/kg/day every 2 weeks. The lesions resolved within 3 weeks and there were no relapses. Adalimumab and infliximab may be utilized for severe refractory patients.
Acanthosis Nigricans
Acanthosis nigricans (AN) is a nonspecific reaction pattern that may accompany obesity; diabetes; excess corticosteroids; pineal tumors; other endocrine disorders ( Table 26.1 ); multiple genetic variants; drugs such as nicotinic acid, estrogens, and corticosteroids; and adenocarcinoma. AN is classified into malignant and benign forms ( Table 26.2 ).
| Metabolic Diseases | Endocrine States | Syndromes |
|---|---|---|
| Insulin-Resistant
|
|
Hyperandrogenic States
|
| Obesity-associated |
|
| Medication-associated |
|
| Syndromic |
|
| Autoimmune |
|
| Acral (acral acanthotic anomaly) |
|
| Unilateral (nevoid acanthosis nigricans) |
|
| Familial |
|
| Malignant |
|
| Mixed-type |
|
Clinical Characteristics.
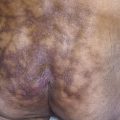
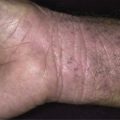
In all cases the disease presents with symmetric, brown thickening of the skin. In time the skin may become quite thickened as the lesion develops a leathery, warty, or papillomatous surface ( Fig. 26.3 and Table 26.2 ). The lesions range in severity from slight discoloration of a small area to extensive involvement of wide areas. The most common site of involvement is the axilla ( Fig. 26.4 ), but the changes may be observed in the flexural areas of the posterior neck ( Fig. 26.5 ) and groin, along the belt line, over the dorsal surfaces of the fingers, in the mouth, and around the areolae of the breasts ( Fig. 26.6 ) and umbilicus. During the disease process, there is papillary hypertrophy, hyperkeratosis, and an increased number of melanocytes in the epidermis.
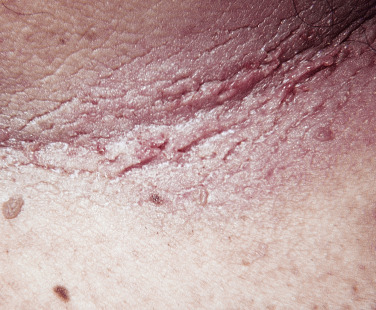
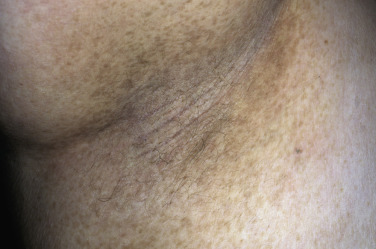
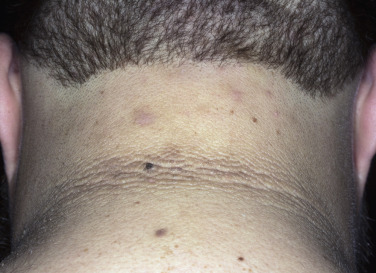
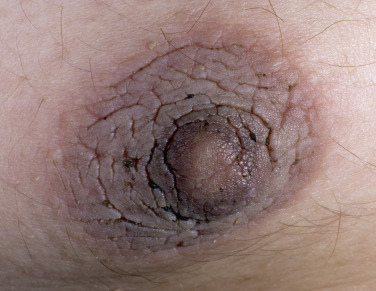
Benign Acanthosis Nigricans.
The majority of cases are idiopathic and are associated with obesity; this process is referred to as pseudo–acanthosis nigricans . Most patients with AN have either clinical or subclinical insulin resistance (IR). There is a high prevalence of AN in obese adults. There is a positive correlation between the development of AN and the severity of the obesity. AN is a common finding in overweight youth. Nearly 40% of Native American teenagers, 13% of black people, 6% of Hispanic people, and less than 1% of white, non-Hispanic children between the ages of 10 and 19 have AN. AN identifies a subgroup within an ethnic group that has the highest insulin concentration, the most severe IR, and the highest risk for the development of type 2 diabetes.
The interaction between excessive amounts of circulating insulin with insulin-like growth factor receptors on keratinocytes may lead to the development of AN.
In rare instances AN may occur as an autosomal-dominant trait with no obesity, associated endocrinopathies, or congenital abnormalities; it may appear at birth or during childhood and is accentuated at puberty.
Drug-Induced Acanthosis Nigricans.
Drug-induced AN has occurred with the use of nicotinic acid, systemic corticosteroids, estrogen, insulin, niacin, oral contraceptive pills, pituitary extract, triazinate, methyltestosterone, fusidic acid, protease inhibitors, diethylstilbestrol, melanocyte-stimulating hormone preparation.
Endocrine Syndromes With Acanthosis Nigricans.
In a large and heterogeneous group of conditions, insulin action at the cellular level is markedly reduced (see Table 26.1 ). AN appears to represent a cutaneous marker of tissue IR, irrespective of its cause (antibodies to the insulin receptor or congenital or acquired defects of receptor or postreceptor function). These patients may not require insulin therapy, and many do not have diabetes. For patients without diabetes, IR is established by the documentation of high levels of circulating insulin or by the observation of an impaired response to exogenous insulin. Prolonged hypersecretion of insulin may lead to pancreatic exhaustion, glucose intolerance, and type 2 diabetes.
Two syndromes of IR and AN are of special interest. Type A syndrome, also called the HAIR-AN syndrome, is characterized by hyperandrogenemia (HA), extreme IR, and AN occurring in the absence of obesity or lipoatrophy. It is distinguished from type B IR by a lack of antibodies to the insulin receptor or other evidence of autoimmune disease. It is familial and affects mainly black women, who have the onset of AN in infancy or childhood.
Treatment.
Lesions are usually asymptomatic and do not require treatment. Reducing thicker lesions in areas of maceration may decrease odor and promote comfort. A 12% ammonium lactate cream, applied as needed, may soften lesions. Retinoic acid (Retin-A cream or gel) applied each day, or less often if irritation occurs, is effective. Calcipotriene (calcipotriol) is thought to decrease keratinocyte proliferation and may be applied twice daily for several months.
Malignant Acanthosis Nigricans.
The cases of greatest concern are those originating in nonobese adult patients. These cases may result from secretion of tumor products with insulin-like activity or transforming growth factor alpha, which stimulates keratinocytes to proliferate. These patients must be evaluated for internal malignancy. The stomach is the most common site for the tumor, but cancer in several other areas has been reported. Malignant AN has a different clinical appearance. Lesions develop rapidly and tend to be more severe and extensive. Hyperpigmentation is prominent and is not limited to the hyperkeratotic areas. Mucous membrane involvement and thickening of the palms and soles occur more frequently. Itching is common. The presence of AN in conjunction with tripe palms and the sign of Leser–Trélat is highly suggestive of an internal malignancy. In approximately one third of patients, the skin lesions precede the clinical manifestations of cancer, and in several cases they have disappeared with successful removal of the tumor. A recurrence of AN may mark the recurrence or metastasis of the previously treated cancer. In patients with malignant acanthosis, chemotherapy may relieve many of the distressing cutaneous symptoms.
Xanthomas and Dyslipoproteinemia
The plasma lipids and lipoprotein levels are under the control of a number of genetic and environmental influences. Abnormalities in a number of these lipids or subfractions result in dyslipoproteinemias and xanthomas. Xanthomas are lipid deposits in the skin and tendons that occur secondary to a lipid abnormality. These localized deposits are yellow and are frequently very firm. Although certain types of xanthomas are characteristic of certain lipid abnormalities, none is absolutely specific because the same form of xanthomas occurs in many different diseases; further investigation is always required. The molecular defect of various lipid disorders is now known; however, the classification and diagnosis are still based on history and clinical presentation ( Table 26.3 ).
| Type | Clinical Characteristics | Associated Lipid Abnormality |
|---|---|---|
| Xanthelasma | Inner or outer canthus; plane or papular | No lipid abnormality; increased frequency of apoE-ND phenotype and hyperapobetalipoproteinemia, type II * |
| Eruptive | Crops of discrete yellow papules on an erythematous base on buttocks, extensor aspects of elbows and knees; lesions clear when triglycerides return to normal | Indicative of hypertriglyceridemia and seen with types I, II, IV, and rarely III and diabetes mellitus |
| Plane | Palms and palmar creases, eyelids, face, neck, chest | Biliary cirrhosis, type III; reported in types II, IV |
| Tuberous | Lipid deposits in dermis and subcutaneous tissue; plaque-like or nodular; frequently found on elbows or knees | Hypertriglyceridemia (familial or acquired), types II and III; biliary cirrhosis |
| Tendinous | Nodules involving elbows, knees, Achilles tendon, and dorsum of hands and feet | Indicates hypercholesterolemia, type II; occasionally type III |
Pathophysiology.
The liver secretes lipoproteins, which are particles composed of various combinations of cholesterol and triglycerides. These particles are made water soluble to facilitate transport to peripheral tissues by polar phospholipids and 12 different specific proteins termed apolipoproteins. The apolipoproteins also serve as cofactors for plasma enzymes and interact with cell surface receptors. Lipoproteins are divided into five major classes: chylomicrons, very-low-density lipoproteins (VLDL), intermediate-density lipoproteins (IDL), low-density lipoproteins (LDL), and high-density lipoproteins (HDL). LDL and HDL have each been divided into two subfractions.
Classification: Primary Versus Secondary Hyperlipoproteinemia.
Dyslipoproteinemias are categorized as primary or secondary. Primary conditions ( Table 26.4 ) are genetically determined and were grouped by Fredrickson into five or six types on the basis of specific lipoprotein elevations. This classification recognizes elevations of chylomicrons (type I), VLDL or pre-beta lipoproteins (type IV), “broad beta” disease (or type III hyperlipoproteinemia), beta lipoproteins (LDL) (type II), and elevations of both chylomicrons and VLDL (type V). In addition, the combined elevations in pre-beta (VLDL) and beta (LDL) lipoproteins were recognized as type IIb hyperlipoproteinemia. This older classification still provides a useful conceptual framework. It does not, however, include HDL cholesterol nor does it differentiate severe monogenic lipoprotein disorders from the more common polygenic disorders. The World Health Organization has classified lipoprotein disorders on the basis of arbitrary cut-points, but the traditional classification will be used in this text.
| Phenotype | Lipoprotein at Increased Concentration | Cholesterol Concentration | Triglyceride Concentration | Dermatologic Lesion(s) |
|---|---|---|---|---|
| I | Chylomicrons | + | ++++ | Eruptive xanthomas |
| IIa | LDL | ++++ | + | Tendon, tuberous, and intertriginous xanthomas; xanthelasma, plane |
| IIb | VLDL and LDL | ++++ | ++ | Tendon, tuberous, and intertriginous xanthomas; xanthelasma, plane |
| III | IDL | +++ | +++ | Tuberoeruptive, tuberous, plane (palmar creases) |
| IV | VLDL | + | +++ | Eruptive xanthomas |
| V | Chylomicrons and VLDL | ++ | ++++ | Eruptive xanthomas |
Secondary hyperlipoproteinemias occur as a result of another disease process that can induce symptoms ( Box 26.1 ), lipoprotein changes, and xanthomas that mimic the primary syndromes. Diagnosis should be made as follows:
- 1.
Determine the type of xanthoma.
- 2.
Measure fasting blood levels of cholesterol, triglycerides, and HDL, VLDL, and LDL.
- 3.
Rule out secondary diseases (see Box 26.1 ). The diagnosis of primary hyperlipoproteinemia is one of exclusion.
- a.
Thyroid, liver, renal function tests
- b.
Glucose tolerance tests
- c.
Complete blood count (CBC); serum and urine immunoelectrophoresis
- d.
Chest X-ray film, bone marrow aspiration
- e.
Antinuclear antibodies (ANAs) testing
- a.
Hypercholesterolemia
Nephrotic syndrome
Hypothyroidism
Dysgammaglobulinemia
Acute intermittent porphyria
Obstructive liver disease
Combined Hyperlipidemia
Nephrotic syndrome
Hypothyroidism
Glucocorticoid excess/Cushing disease
Diuretics
Uncontrolled diabetes
Alcohol
Estrogens
Beta-adrenergic blocking agents
Isotretinoin (13- cis -retinoic acid)
Hypertriglyceridemia
Diabetes mellitus
Uremia
Sepsis
Obesity
Systemic lupus erythematosus
Dysgammaglobulinemia
Glycogen storage disease, type I
Lipodystrophy
Drugs
Alcohol
Estrogens
Beta-adrenergic blocking agents
Isotretinoin (13- cis -retinoic acid)
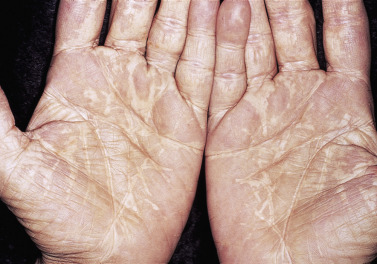
Xanthelasma and Plane Xanthomas.
Plane xanthomas occur in several areas of the body and are flat or slightly elevated ( Figs. 26.7 and 26.8 ). Xanthelasma is the most common form (see Fig. 26.7 ). Xanthelasma can be associated with familial hypercholesterolemia, phenotype IIa or IIb, but 50% of the patients have normal cholesterol levels. Longevity studies have shown that xanthelasma, with or without hypercholesterolemia, is a risk factor for death from atherosclerotic disease. Further study of these patients with normal levels of cholesterol and triglycerides often reveals elevated LDL and VLDL levels and decreased HDL level. This profile is found for patients who have a high risk of atherosclerotic cardiovascular disease. Premature carotid atherosclerosis is observed in some patients with normolipidemic and hyperlipidemic xanthelasma. Patients with xanthelasma should be considered to have an increased risk of cardiovascular disease independent to the level of plasma lipids. It may be that all patients with xanthelasma have an increased risk for atherosclerosis.
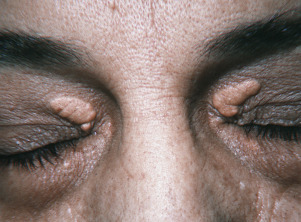
Trichloroacetic acid (TCA) is commonly used for cosmetic treatment. Papulonodular lesions required an average of two applications with 100% TCA, three with 70% TCA, and four with 50% TCA. Flat plaques responded to an average of one, two, and three sittings with 100%, 70%, and 50% TCA, respectively. Macular lesions responded to only one application of all strengths of TCA applied. Hypopigmentation is the most common side effect, followed by hyperpigmentation. Scarring is a minor problem.
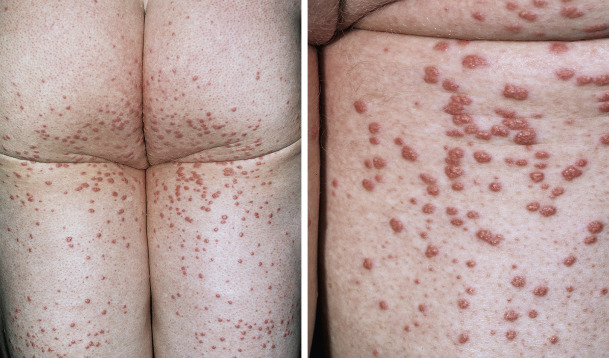
Eruptive Xanthomas.
These are yellow, 1- to 4-mm papules with a red halo around the base. They appear suddenly in crops on extensor surfaces of the arms, legs, and buttocks and over pressure points ( Fig. 26.9 ). Lesions clear rapidly when serum lipid levels are lowered.
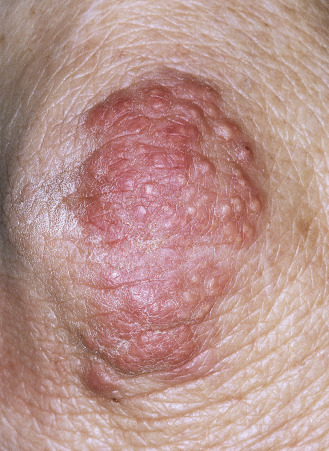
Tuberous Xanthomas.
These are slowly evolving yellow papules, nodules, or tumors that occur on the knees, elbows, and extensor surfaces of the body and the palms ( Fig. 26.10 ).
Tendinous Xanthomas.
These smooth, deeply situated nodules are attached to tendons, ligaments, and fascia. They are most often found on the Achilles tendons and the dorsal aspects of the fingers.
Regression of Xanthomas.
Certain xanthomas disappear with treatment. The eruptive and palmar xanthomas can regress rapidly. The eruptive type of tuberous xanthomas can disappear. Tendinous xanthomatous lesions tend to persist.
Neurofibromatosis
The neurofibromatoses (NF) comprise at least two autosomal-dominant disorders with an incidence of approximately 1 in 3000. These diseases have tumors surrounding nerves. Riccardi classified NF into eight types. Neurofibromatosis 1 (NF1) is the most common and is characterized by congenital lesions of the skin, central nervous system, bone, and endocrine glands. The cardinal features of the disorder are café-au-lait macules (CALMs), axillary freckling, cutaneous neurofibromas, and iris hamartomas (Lisch nodules). Common complications include learning disability, scoliosis, and optic gliomas. Neurofibromatosis 2 (NF2) is characterized by bilateral acoustic neuromas and other nerve tumors. Skin and other systemic manifestations are minimal or absent. Café-au-lait macules, freckling, and neurofibromas localized to a segment of the body are called segmental neurofibromatosis (NF5). The NF1 gene is located on chromosome 17 and the NF2 gene is found on chromosome 22.
Neurofibromatosis 1
NF1 is a disorder of neural crest–derived cells characterized by the presence of café-au-lait macules, multiple neurofibromas, and Lisch nodules (pigmented iris hamartomas); there are several other less common features. There is considerable variation of manifestations within the same family. It occurs in approximately 1 of every 3500 births and affects both genders with equal frequency and severity. Neurofibromatosis is one of the most common genetic condition in humans; at least half of the cases represent new mutations in the neurofibromin gene.
Clinical Manifestations
Café-au-Lait Macules.
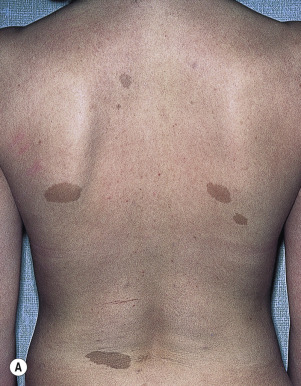
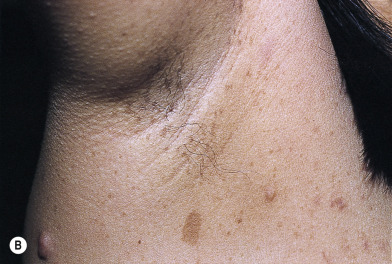
Café-au-lait macules are light-colored to brown macules (see Chapter 19 ). The criteria for establishing the diagnosis with reference to the number and size of CALMs are listed in Box 26.2 . The macules are present in virtually every patient with NF, usually at birth, but they may not appear for months. Their size and number increase with age ( Fig. 26.11A ). Intertriginous freckling, a pathognomonic sign, may occur in the axillae, inframammary region, and groin ( Fig. 26.11B ). Café-au-lait macules alone are not absolutely diagnostic of NF1, regardless of their size and number.
Six or more café-au-lait macules
- •
Greater than 5 mm in greatest diameter in prepubertal individuals
- •
Greater than 15 mm in greatest diameter in postpubertal individuals
- •
One plexiform neurofibroma or two or more neurofibromas of any type
Axillary or inguinal freckling
Two or more Lisch nodules (iris hamartomas)
Optic glioma
Distinctive osseous lesion (e.g., sphenoid dysplasia or tibial pseudoarthrosis)
First-degree relative with NF1 as defined by the above criteria