Cryoglobulinemia
|
Cryoglobulinemia may be a symptomless phenomenon that is incidentally found, or it may produce a clinical syndrome involving the skin as a primary organ. In a historic large-scale examination, 11% of patient sera were shown to have cyoglobulinemia, but detection rates vary greatly with the patient population studied.1 Reliable figures on the incidence or prevalence of cryoglobulinemia or symptomatic cryoglobulinemic syndrome are lacking. The epidemiologic study is hampered by factors such as selection bias from the referral of patients to tertiary care hospitals, by the heterogeneity of clinical presentations inclusion criteria, and by the different geographic distribution of underlying etiologies such as HCV infection. There is a geographic predominance of cryoglobulinemia in Southern Europe as compared to Northern Europe or the United States that might be explained by this latter fact. The frequency distribution of different cryoglobulinemia subtypes among all types of cryoglobulinemia is approximately type I 25%, type II 25%, type III 50%, making the mixed cryoglobulinemias (types II and III) the most abundant form.2 The vast majority (>90%) of mixed cryoglobulinemias occurs in association with chronic HCV infection. Thus, HCV-associated mixed cryoglobulinemia is the form among all cryoglobulinemias that has been studied best. On average, 30% of chronically HCV-infected subjects develop cryoglobulinemia, and this proportion can go up to 90% when patients with very longstanding disease are examined.3–5 Development of cryoglobulins is more frequent with HCV genotype 1 as compared to genotypes 2 and 3.6 However, on average, the symptomatic cryoglobulinemic vasculitis syndrome is present in only 2% to 5% of chronically HCV-infected patients7 though figures as high as 15% have been proposed.8,9 With a conservatively estimated prevalence of chronic HCV infection of ∼1% in mind,10 the calculated prevalence of HCV-associated cryoglobulinemic vasculitis in the USA would be 20–50 per 100,000. The true prevalence is probably on the lower end of this estimation. Given the increasing proliferation of HCV infection worldwide, rising figures might be expected on a global perspective.11 The prevalence of “essential mixed cryoglobulinemia,” i.e., mixed cryoglobulinemia without an identifiable cause, has been estimated to be 1:100,000 in former studies, with a female-to-male ratio of 3:1. However, this number is difficult to interpret since many cases that are nowadays recognized to be associated with HCV or other viruses were formerly attributed to be “essential.”12
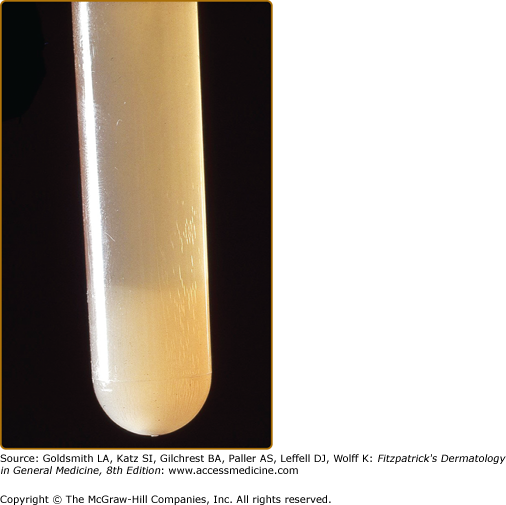
In 1933, Wintrobe and Buell first noted the in vitro phenomenon of cryoprecipitation, i.e., the cold-induced precipitation of plasma or serum proteins that is reversible upon rewarming at 37°C (98.6°F).13 In 1947, Lerner and Watson described immunoglobulins and mixtures of immunoglobulins with other proteins that precipitate in the cold and called them cryoglobulins.1 In 1974, Brouet introduced the classification of cryoglobulins based on molecular properties that is currently used.2 Cryoglobulinemia describes the presence of cryoglobulins in a patient’s serum (see Fig. 169-1). It can be asymptomatic, meaning that its presence usually goes undetected outside academic study screening protocols, or it can cause occlusive vasculopathy or the so-called cryoglobulinemic syndrome, which is characterized by immune-complex deposition causing vasculitis that involves the skin and mainly neural and renal tissues.
The classification scheme of Brouet, estimated frequency and composition of cryoglobulins is depicted in Table 169-1. Type-I cryoglobulins consist of a single monoclonal immunoglobulin (Ig), typically IgM, less commonly IgG or IgA or free Ig light chains (Bence Jones proteins). Complement components are not routinely found in type I cryoprecipitates. Type I cryoglobulins are often present in substantial amounts, ranging from 1 to 30 mg/mL. The large molecular size of monoclonal IgM and other molecular characteristics, such as absence of sialic acid moieties, deficient carbohydrate side chains, and weak noncovalent factors may predispose these Igs to precipitation.14 Type I cryoglobulinemia is associated with hematologic disorders, such as Waldenström macroglobulinemia, multiple myeloma, or lymphoproliferative diseases (e.g., B-cell lymphoma).
Type of Cryoglobulinemia (Estimated Frequency) | Composition of Cryoprecipitates | Disease Associations |
|---|---|---|
Type I (25%) | Monoclonal IgM (sometimes IgG, IgA), immunoglobulin light chain, complexed to other proteins | Haematological disorders
|
Type II (25%) | Combination of monoclonal (usually IgM with rheumatoid factor activity) and polyclonal (usually IgG) | Chronic infection
Autoimmune diseases
Haematological disorders
|
Type III (50%) | Polyclonal Igs | Chronic infection
Autoimmune diseases
|
Type II–III (frequency unknown) | Oligoclonal IgM, intermediate state between the entirely polyclonal type III and the monoclonal, polyclonal type II | Chronic infection (e.g., HCV) Autoimmune diseases Lymphoproliferative diseases Chronic liver disease Proliferative glomerulonephritis |
Type II and type III cryoglobulins are immune complexes composed of polyclonal IgGs and mono- or polyclonal IgMs. Type II and type III cryoglobulins are typically present in relatively small amounts and generally result from chronic inflammatory states. Type II and type III cryoglobulins may fix complement.
Type II cryoglobulins represent a mixture of two Ig components: polyclonal Igs are associated with a monoclonal Ig that exhibits rheumatoid factor (RF) activity. Typically, a monoclonal IgM RF is complexed with a polyclonal IgG. HCV infection is the classical underlying disease.
A mixture of polyclonal Igs or polyclonal Ig-nonimmunoglobulin cryoprecipitates results in detection of type III cryoglobulins. Polyclonal IgM–IgG cryoglobulins with complement as an integral component are the most frequent scenario in type III cryoglobulinemia that is commonly associated with HCV infection or connective tissue diseases.
A new type of cryoglobulins, called type II–III cryoglobulin, containing polyclonal IgG associated with a mixture of polyclonal and monoclonal (oligoclonal) IgM has recently been described.15 This type of cryoprecipitate describes an intermediate, developing state between types III and II, suggesting a continuous transition from a purely polyclonal to a partially monoclonal composition by a process of successive clonal selection.
Types II and III cryoglobulinemias are classically referred to as “mixed cryoglobulinemias”. Circulating mixed cryoglobulins are commonly detected in a great number of infectious or systemic disorders (see Table 169-1). The term “essential” is originally reserved for instances of mixed cryoglobulinemia in the absence of a well-defined underlying disease and, given the striking association between mixed cryoglobulinemia and HCV, is actually referred to only a minority of cryoglobulinemic patients.
Prevalence of serum anti-HCV antibodies and/or HCV RNA in cryoglobulinemic patients ranges from 70% to 100%. Type II cryoglobulinemia is more strongly associated with HCV than type III (90% vs. 70%, respectively). Presence of cryoglobulins increases with duration of HCV infection: cryoglobulins are found in 55%–90% of patients with longstanding infection. However, overt cryoglobulinemic syndrome develops in only 2%–5% of these cases (see also Section “Epidemiology”). The precise role of viral or host factors contributing to this discrepancy remains largely unknown. Specific HCV genotypes or distinct HLA subtypes, like HLA-DR11 or HLA-DR6 may predispose to extrahepatic systemic manifestations of cryoglobulinemia.16,17 Pathogenesis of mixed cryoglobulinemia is probably a multifactorial and multistep process. Viral HCV antigens exert a chronic stimulus on the host immune system, resulting in specific immune dysregulatory mechanisms with B-cell proliferation and autoantibody production. HCV-related B-cell lymphoproliferative disorders comprise a spectrum of disease, ranging from asymptomatic clonal B-cell expansions to pathogenic cryoglobulinemia and lymphoma.18 From that perspective, HCV-associated mixed cryoglobulinemia is a benign lymphoproliferative B-cell disease with a potential for subsequent development of B-cell lymphoma.19,20 Development of HCV-associated cryoglobulinemic vasculitis syndrome is associated with longstanding infection, old age, type II cryoglobulins, higher cryoglobulin levels, and clonal B-cell expansion.21,22
The group of non-HCV-induced mixed cryoglobulinemias is small. The following associations have been described: An association of cryoglobulinemia with various other infectious viral agents, for example, hepatitis B virus, cited as a major causative agent,23 and HIV with or without HCV coinfection has also been demonstrated.24 Parvovirus B19 has been implicated in a mild form of cryoglobulinemic syndrome.25 Another important association is the one with Sjögren syndrome. According to one study, 16% of patients with primary Sjögren syndrome had cryoglobulins, and of those, 56% had cryoglobulinemic syndrome.26,27 A study by the same group found cryoglobulinemia in 25% of patients with systemic lupus erythematosus.28
In almost all cases, the possibility of a diagnosis of cryoglobulinemia or cryoglobulinemic syndrome is primarily suggested by the presence of purpura of the distal extremities or by acral cyanosis and necrosis, depending on the potential presence of type I or types II and III cryoglobulinemia. Alternatively, the potential diagnosis may be suggested by the presence of a candidate underlying disease. A targeted review of systems and subsequent clinical examination will guide the physician towards the entire picture of organ involvement or differential diagnoses. The laboratory panel must include parameters relevant to both cryoglobulinemia and important differential diagnoses. Box 169-1 presents an algorithm for the diagnostic approach to the patient, including laboratory testing. eTable 169-1.1 summarizes the symptoms, signs, cutaneous, extracutaneous and laboratory findings of cryoglobulinemia. Of note, there are still only preliminary diagnostic criteria for the classification of mixed cryoglobulinemic patients (see eTable 169-1.2).
HISTORY AND EXAMINATION (INCLUDING OPTIONAL TECHNICAL METHODS) |
Ask for
|
Look for
|
Technical examination (optional)
|
MANDATORY LABORATORY TESTING IN CRYOGLOBULINEMIA
|
HISTOLOGY
|
|
Proposed Criteria for the Classification of Mixed Cryoglobulinemia (MC) Patients | |||
Criteria | Serological | Pathological | Clinical |
Major | Mixed cryoglobulins Low C4 | Leukocytoclastic vasculitis | Purpura |
Minor | Rheumatoid factor + HCV + HBV + | Clonal B-cell infiltrates (liver and/or bone marrow) | Chronic hepatitis MPGN Peripheral neuropathy Skin ulcers |
Definite mixed cryoglobulinemia syndrome | |||
(a) Serum mixed cryoglobulins (± low C4) + purpura + leukocytoclastic vasculitis (b) Serum mixed cryoglobulins (± low C4) + two minor clinical symptoms + two minor serological/pathological findings | |||
Essential or secondary mixed cryoglobulinemia Absence or presence of well-known disorders (infectious, immunological, or neoplastic) | |||
Cutaneous lesions are the most frequent manifestations (Fig. 169-2). Type I cryoglobulinemia per se is often asymptomatic and cutaneous signs are related to hyperviscosity and/or thrombosis that induce ischemic vasculopathy presenting as Raynaud phenomenon, digital ischemia, livedo reticularis, or purpura. Type II and III cryoglobulinemic vasculitis is mediated by deposition of antigen–antibody complexes in small- and medium-sized arteries, leading to inflammation of vessel walls. Intermittent orthostatic palpable purpura, frequently observed late in the afternoon when highest cryoglobulin concentrations are present, is the most common presentation and particularly affects the lower extremities.29 Dimension and diffusion of purpuric lesions can vary from sporadic isolated petechias or erythematous macules to severe vasculitic lesions with ulcerations. Leukocytoclastic vasculitis is the histopathological hallmark of mixed cryoglobulinemia and is easily detectable by skin biopsy (see Chapter 163 see also eFig. 169-2.1). Nail-fold capillary abnormalities are common and include dilatation, altered orientation, capillary shortening, and neoangiogenesis.
eFigure 169-2.1
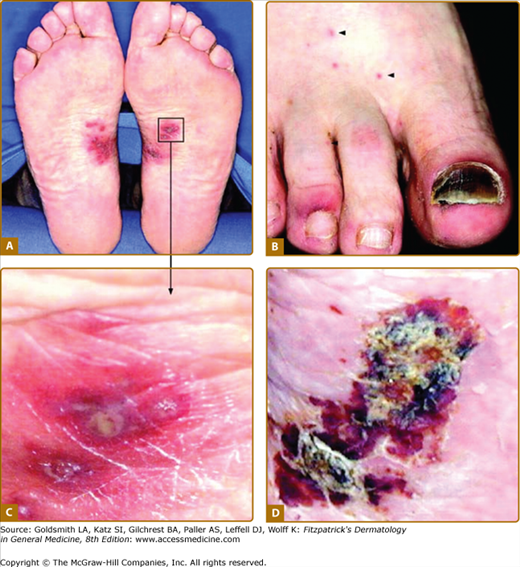
Dermal findings in a patient with type II/III cryoglobulinemia associated with chronic alcoholic liver disease in the absence of viral hepatitis. A. Symmetrical purpuric papular lesions on the medial aspect of the foot soles. B. Petechial macular lesions on the dorsal aspect of the foot on presentation. C. Magnification of the boxed area from panel A. D Partial healing of the foot sole lesion after 8 weeks of oral steroid treatment. Presence of type II/III cryoglobulin in the absence of other pathologic immune serologies or evidence for neoplasm or internal organ involvement confirmed the diagnosis of isolated dermal cryoglobulinemic vasculitis in this patient. (Reproduced with permission from Braun GS et al: Cryoglobulinaemic vasculitis: classification and clinical and therapeutic aspects. Postgrad Med J 83:87-94, 2007.)
Renal involvement is a serious complication, typically manifests early in the course of the disease and can present as a broad range of clinical findings, including hematuria with or without renal insufficiency (41%), isolated proteinuria, nephrotic syndrome (21%) or acute nephritic syndrome.30 Chronic renal insufficiency without significant renal abnormalities and acute renal failure are less common. The incidence of renal disease in cryoglobulinemia varies from 5% to 60% and is typically immune complex mediated (type II and III), but may also occur secondary to thrombosis (type I) (for details see eTable 169-1.3).
Cryoglobulinemia | Frequency of renal involvement | Associated Kidney Disease | Pathogenesis | Reference |
|---|---|---|---|---|
Type I | Extremely rare | Glomerulonephritis | ||
Rare | Tubular damage (Fanconi syndrome)—depositions in the glomerular basement membrane and the mesangium | Plasma cell dyscrasias associated light chain and amyloid depositions | ||
Type II | Most common (∼50%) | MPGN Ia (∼85%)a Mesangioproliferative GN (∼7%)a | Fibronectin specific monoclonal IgM leading to glomerular leukocyte attraction and damage | |
Type III | Less common (∼10%) | MPGN I (∼85%)b Mesangioproliferative GN (∼7%)b | Unknown, possibly like cryoglobulinemia type II |
Neurological manifestations, typically affecting the sensory peripheral nervous system secondary to epineural vasculitis frequently complicate the clinical course. Patients typically describe paresthesias with burning symptoms in the lower limbs, often with nocturnal exacerbation, leading to severely compromised quality of life. Electromyographic and nerve conduction studies demonstrated peripheral neuropathy in up to 80% of patients with mixed cryoglobulinemia, but many symptom-based demographic studies report prevalence of only 5% to 45%. Peripheral neurologic disease manifests as a progressive, chronic distal mild sensory neuritis and only rarely as acute mononeuritis. Clinically apparent central nervous system dysfunction is rare.
Musculoskeletal complaints, typically arthralgias and myalgias, are described in more than 70% of persons with cryoglobulinemia, predominantly in type II and III disease. The association of cryoglobulinemia with palpable purpura, arthralgia and myalgia was referred to as Meltzer triad in the late 1960s. Arthralgias classically affect the proximal interphalangeal and metacarpophalangeal joints of the hands, the knees, and ankles. Clear clinical signs of myositis or arthritis are rare.
Abnormal liver function tests, hepatomegaly including signs of cirrhosis or abnormal histological results in liver biopsy were reported in up to 90%. Also, thyroid disorders and diabetes mellitus seem to be associated with cryoglobulinemia.39
Unspecific abdominal pain can affect 2% to 22%. Intestinal vasculitis of the small mesenteric vessels leading to acute abdomen has been reported.
Approximately 40% of patients are symptomatic with dyspnea, cough, or pleuritic pain. Pulmonary function tests often reveal evidence of small airways disease and chest radiographs sometimes show interstitial infiltrates or signs of subclinical alveolitis. Severe pulmonary disease, for example, bronchiolitis obliterans organizing pneumonia (BOOP) or pulmonary vasculitis are very uncommon.
Hyperviscosity due to high levels of monoclonal cryoglobulins, typically seen in Waldenström macroglobulinemia and less frequently in multiple myeloma, can induce microcirculation impairment and reduced platelet function, leading to skin and mucosal bleeding. A variety of neurologic symptoms have been reported in hyperviscosity syndrome: blurring or loss of vision, headache, vertigo, nystagmus, dizziness, sudden deafness, diplopia, ataxia, confusion, dementia, disturbances of consciousness, stroke, seizures, somnolence or coma. A characteristic retinal venous engorgement (“sausaging”) on funduscopic inspection can serve as a diagnostic clue.
In clinical practice, cryoglobulin testing is underutilized, most probably due to the expenditure of time and stringent temperature requirements.
Serum must be obtained in warm tubes (37°C) in the absence of anticoagulants. After clotting and centrifugation at 37°C, the separated serum is stored at 4°C and inspected daily for a precipitate. Type I cryoglobulins tend to precipitate within the first 24 hours (at concentrations >5 mg/mL), whereas type III cryoglobulins may require 7 days (see Fig. 169-1). For calculation of cryocrit (volume of packed cryoglobulins as percentage of original serum volume), the cryoprecipitate has to be spun in a graded (e.g., Wintrobe) tube. A cryocrit ≥2% is considered to be positive. Cryocrit levels usually do not correlate with severity and prognosis of disease.
Some authors recommend proof of reversibility of the cryoprecipitate by rewarming an aliquot at 37°C for 24 hours.12 For phenotyping and identification of cryoglobulin components specific immunologic assays are performed at 37°C.
Clinical diagnosis of hyperviscosity can be established by measuring serum viscosity with an Oswald-type viscometer. Reference serum viscosity, measured as flow time through the viscometer of the patient’s serum divided by that of water or saline, is between 1.4 and 1.8, while most symptomatic patients have values between 5 and 8. Again, clinical manifestations are often not proportional to serum viscosity.
As hypocomplementemia (with the typical pattern of low or undetectable C4 and normal or relatively normal C3 levels) occurs in up to 90% of patients with mixed cryoglobulinemia, C3 and C4 levels, usually measured in nephelometric immunoassays, should be routinely determined. A sudden increase in complement C4, raised to abnormally high levels can be observed in some patients developing a B-cell lymphoma.40
RF is often positive in type II and III cryoglobulinemia. Testing for antinuclear antibodies (ANAs, SS-A, SS-B) and other autoantibodies (e.g., anti-smooth-muscle Abs) is indicated upon clinical suspicion of underlying systemic connective tissue disease (e.g., SLE, Sjögren syndrome). Although the immunofluorescence ANA assay is the current diagnostic gold standard, titer and specificity of ANAs may vary considerably.
Particularly in patients with HCV-associated mixed cryoglobulinemia serum levels of IL-1β, IL-6, and TNF-α are significantly elevated.41 Increased levels of the chemokines CXCL10 and CXCL13, also known as BCA-1 (B-cell-attracting chemokine-1), seem to be associated with active vasculitis.42,43
Vasculitic purpura is histologically characterized by dermal leukocytoclastic vasculitis that extends variably to the subcutaneous interstitial space (Cross-reference leukocytoclastic vasculitis). For detailed histological findings in leukocytoclastic vasculitis (e.g., composite of inflammatory infiltrates) see Figure X, Section Leukocytoclastic Vasculitis. Deposition of immunoglobulin, complement and HCV-associated proteins in mixed cryoglobulinemia is common. Abnormalities of uninvolved skin include basement membrane alterations and deposits in vessel walls.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


