I. CRANIOFACIAL EMBRYOLOGY AND DEVELOPMENT
A. Skeletal tissues of head and face derive from mesenchyme and cranial neural crest cells
B. Bone formed through both endochondral and intramembranous ossification (Fig. 27-1)
1. Skull development starts at 23 to 26 days of gestation
2. Neurocranium: Develops into calvarium and provides bony encasement around the brain
a. Membranous neocranium: Precursor to cranial vault
i. Paired frontal, parietal, squamosal temporal, and superior occipital bone
ii. *Bone formation through intramembranous ossification (direct ossification of mesenchyme)
b. Cartilaginous neocranium: Precursor to skull base
i. Includes sphenoid, ethmoid, mastoid, petrous portion of temporal bone, and inferior occipital bone
ii. *Bones develop through endochondral ossification (ossification of cartilaginous precursor)
3. Viscerocranium: Precursor to the bones of the facial skeleton
a. *Derived from neural crest cells of the first pharyngeal arch (Meckel cartilage)
i. Maxillary process (dorsal portion of first pharyngeal arch) forms premaxilla, maxilla, zygoma, and squamous temporal bone
ii. Mandibular process (ventral portion of first pharyngeal arch) forms the mandible, malleus, and incus
b. *Second pharyngeal arch (Reichert cartilage) gives rise to stapes, styloid process of the temporal bone and lesser horn and superior body of the hyoid bone
c. Bone formation through intramembranous ossification
C. Cranial sutures
1. Fibrous joints between calvarial bones
2. Metopic, sagittal, coronal, lambdoid, and squamosal (Fig. 27-1)
a. Permit deformational changes (i.e., passage through birth canal)
b. Consist of adjacent osteogenic fronts, interposed mesenchymal tissue, and underlying dura mater
c. Allow for head expansion during development
i. *Primary stimulus for skull growth is brain growth
ii. Brain is 25% of adult size at birth, 50% at 6 months, and 75% at 1 year
iii. Full adult volume by ~2.5 years
3. Fontanelles (infantile “soft spots”) are the confluence of two or more cranial sutures
a. Anterior fontanelle (bregma): Closes around 2 years of age
b. Posterior fontanelle (lambda): Closes around 2 months of age
______________
*Denotes common in-service examination topics
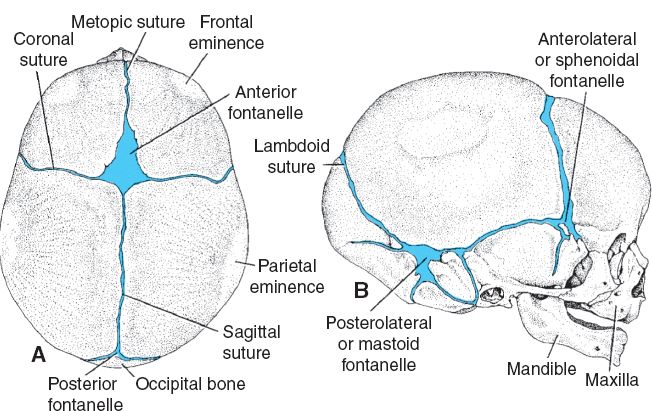
Figure 27-1. Major bones, fontanelles, and cranial sutures of the newborn skull as seen from (A) superior and (B) lateral views. (Modified from Sadler T. Langman’s Medical Embryology. 9th ed. Image Bank. Baltimore, MD: Lippincott Williams & Wilkins; 2003.)

4. Suture fusion sequence
a. Metopic: 3 to 9 months (only suture to obliterate during childhood)
b. Sagittal: 20 to 22 years
c. Coronal: 23 to 24 years
d. Lambdoid: 26 years
D. Sinus development (Table 27-1)
II. CRANIOSYNOSTOSIS
A. Premature fusion of the cranial sutures
B. *Virchow’s law
1. Growth restriction occurs perpendicular to the affected suture
2. Compensatory skull growth occurs parallel to the affected suture
C. Nonsyndromic (primary) craniosynostosis
1. Isolated suture fusion without associated abnormalities
2. Largely sporadic pattern of occurrence (incidence 0.6 in 1,000 live births)
D. Syndromic craniosynostosis
1. Heterogeneous group of disorders marked by premature suture fusion
2. Associated dysmorphic features and congenital abnormalities
3. Genetic heritability patterns (e.g., autosomal dominance, autosomal recessive, and X-linked)
4. Linked to specific gene mutations in some cases (see below)
E. Secondary craniosynostosis: Premature suture fusion due to other disease processes
1. Hyperthyroidism
2. Idiopathic hypercalcemia
3. Rickets
4. Microcephaly
5. Mucopolysaccharidoses
6. Hematologic disorders (thalassemia, polycythemia vera, and sickle cell)
7. Iatrogenic (e.g., after shunt placement for hydrocephalus)
F. Diagnosis, workup, and consultations
1. History
a. Abnormal head contour
b. Sleep disturbances
c. Regression or failure to meet developmental milestones
2. Physical exam
a. Palpable ridge along synostotic sutures
b. Lack of movement along sutures with palpation
c. Dysmorphic facial features or facial asymmetry
d. Abnormal head circumference when compared with age-predicted norms
e. Poorly defined, absent or bulging fontanelles
f. Fundoscopic examination for papilledema
3. Evaluate for elevated intracranial pressure (ICP)
a. Approximately 10% of single suture synostosis and 40% of patients with multisuture synostosis have elevated ICP
b. Irritability, growth impairment, inconsolability, vomiting, bulging fontanelles, and papilledema
c. Requisite neurosurgical consultation in all confirmed patients
4. Imaging: CT scan
a. Routinely used in diagnosis
b. Three-dimensional reformatting for preoperative planning
c. Evidence of elevated ICP may be manifested as hydrocephalus or luckenschadel (“copper beaten”) skull
5. Genetics evaluation
6. Neuropsychological evaluation to determine baseline cognitive functioning
7. Speech and audiology assessment should be performed to ensure ongoing language acquisition during development
III. NONSYNDROMIC CRANIOSYNOSTOSIS
A. Metopic synostosis
1. Relatively uncommon: <10% of craniosynostosis
2. Deformity: Trigonocephaly
3. Associated findings: Keel-shaped skull with pointed forehead, frontal bossing, bitemporal narrowing, hypotelorism, and recessed superior orbital rims
B. Sagittal synostosis
1. *Most common: >50% of craniosynostoses
2. Male predominance: 4:1 male/female ratio
3. Sporadic with 2% genetic predisposition
4. Deformity: Scaphocephaly (dolichocephaly)
5. Associated findings: Increased AP length of skull, “boat-like” appearance, decreased biparietal width, and frontal and occipital bossing
C. Unilateral coronal (unicoronal) synostosis
1. Second most common: 20% of craniosynostosis
2. Deformity: Anterior plagiocephaly
3. Associated findings: Ipsilateral frontal bone flattening, contralateral compensatory frontal bossing, shortened AP dimension on affected side, anterior displacement of ipsilateral ear, and deviation of nasal tip to contralateral side
4. *Harlequin eye deformity
a. Lack of ipsilateral descent of greater wing of sphenoid during development
b. Pathognomonic for unicoronal synostosis
D. Bilateral coronal (bicoronal) synostosis
1. *Most commonly associated with syndromic craniosynostosis (such as Crouzon’s and Apert syndromes)
2. Deformity: Brachycephaly
3. Associated findings: Frontal bossing with vertical elongation of frontal bones, widening of anterior cranial base, shortened AP skull dimension, occipital flattening, shallow orbits, and hypertelorism
E. Lambdoid synostosis
1. Least common: <3% of craniosynostosis
2. May be unilateral or bilateral
3. Deformity: Posterior plagiocephaly
4. Associated findings: Flattening of ipsilateral occiput, posterior and inferior displacement of ipsilateral ear, and contralateral occipitoparietal bossing
F. Multiple suture synostosis
1. Usually occurs as a feature of syndromic craniosynostosis
2. Dysmorphic features depend on the pattern of involved sutures
3. Pansynostosis: Fusion of all cranial sutures
a. Nonsyndromic pansynostosis
i. Inadequate volume expansion secondary to impaired brain growth
ii. Skull is normocephalic in contour but microcephalic in volume
b. Syndromic pansynostosis
i. Normal brain growth which is constrained by the fusion of all sutures
ii. Kleeblattschädel deformity: Clover leaf skull
a) Ballooning of cranial vertex and squamosal sutures
b) Often requires immediate surgical decompression following birth to avert neurologic compromise
c) May require cerebrospinal fluid (CSF) shunt
d) C-spine must be evaluated for additional abnormalities
IV. DEFORMATIONAL PLAGIOCEPHALY (POSITIONAL HEAD DEFORMITY, OR POSITIONAL PLAGIOCEPHALY)
A. Results from external pressure applied to the pliable fetal or infant skull
B. *Must be differentiated from lambdoid or coronal synostosis (Fig. 27-2)
C. Causes
1. Supine sleeping position: Recommended to decrease the risk of sudden infant death syndrome. This is the most common cause of positional plagiocephaly.
2. In utero compression
3. Vertebral abnormalities
4. Congenital muscular torticollis (often occurs with deformational plagiocephaly)
5. Ocular torticollis: Visual field deficits causing preferential head positioning
D. Treatment specific to the underlying cause
1. Supervised prone positioning (“tummy time”), physical therapy, stretching and possible muscle release for torticollis, head and neck rotation while feeding, encouraging looking to affected side by positioning infant
2. Shaping helmets
a. Fitted to widest skull dimension
b. Compensatory growth occurs due to external forces applied by helmet
c. Must be worn for >23 hours per day
d. Effective if worn for 2 to 3 months or longer
e. Less effective after 18 months of age; early helmeting is much more effective
f. Multiple helmet fittings required as cranial contour improves
V. SYNDROMIC CRANIOSYNOSTOSIS
A. Apert syndrome (Fig. 27-3A)
1. Genetics
a. Autosomal-dominant inheritance but vast majority of cases represent sporadic mutations
b. *FGFR2 mutation (chromosome 10)
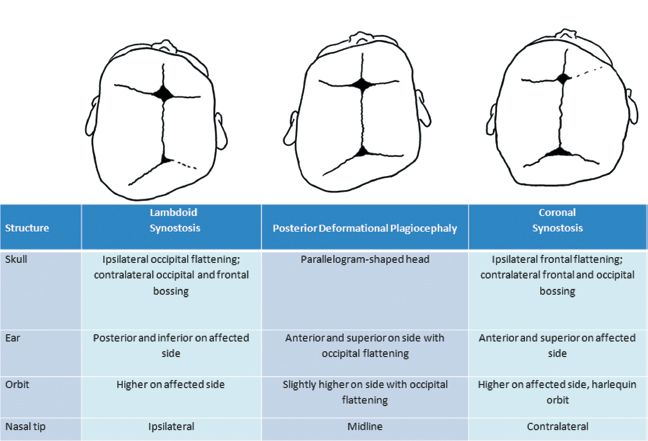
Figure 27-2. Distinguishing features of deformational and synostotic plagiocephaly.
2. Craniofacial features
a. Bicoronal craniosynostosis, turribrachycephaly (short AP skull dimension, wide transverse dimension, and increased vertical excess of skull), orbital hypertelorism, proptosis, midface hypoplasia with class III malocclusion, “parrot beak” nose, high arched palate, occasional cleft palate, and acne
b. Elevated ICP common
3. *Extremities: Severe complex syndactyly of hands and feet in which most or all digits are fused, including phalanges; all interphalangeal joints of fingers are stiff; lack PIP joints; also often have stiffness affecting elbow and shoulder joints; 4 to 5 metacarpal synostosis; and radial clinodactyly of the thumb
4. Mental status: variable
B. Crouzon’s syndrome (Fig. 27-3B)
1. Genetics
a. Autosomal dominant
b. FGFR2 mutation
2. Craniofacial features
a. Coronal and lambdoidal synostosis, turribrachycephaly, exorbitism/proptosis leading to exposure keratitis, midface hypoplasia, class III malocclusion
b. Features less severe than Apert
3. Conductive hearing loss due to cranial base abnormalities
4. *Extremities: normal
5. Mental status: variable
C. Saethre–Chotzen syndrome
1. Genetics
a. Autosomal dominant
b. TWIST-1 mutation
2. Craniofacial features: Asymmetric coronal synostosis, shallow orbits, telecanthus, ptosis of eyelids, midface hypoplasia, deviated nasal septum, low hairline
3. Extremities: Partial syndactyly
4. Mental status: Usually normal
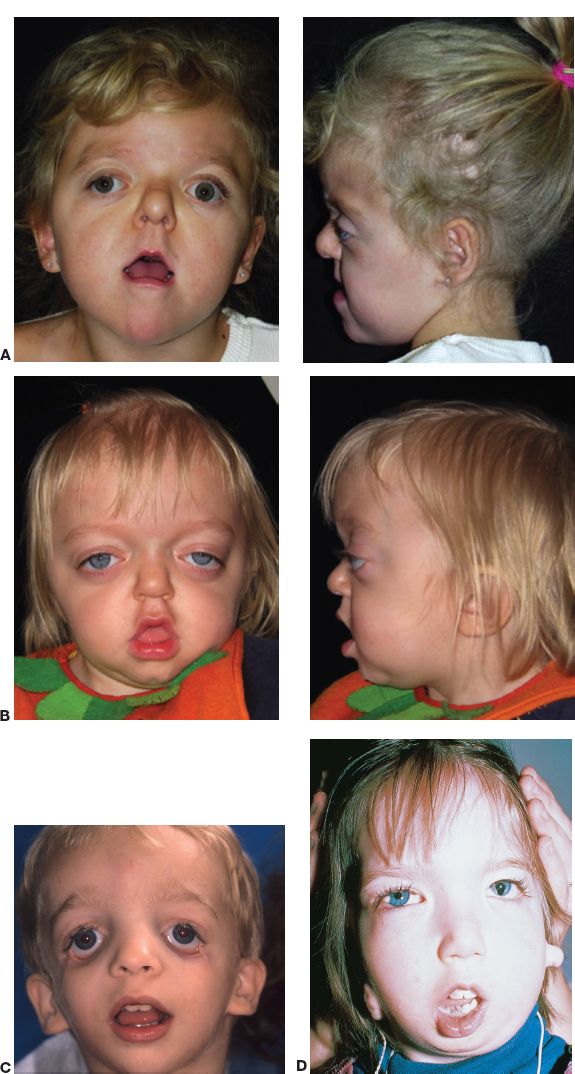
Figure 27-3. A: Apert syndrome. B: Crouzon’s syndrome. C: Treacher–Collins syndrome. D: Goldenhar–Gorlin’s syndrome (From Gold DH, Weingeist TA. Color Atlas of the Eye in Systemic Disease. Baltimore, MD: Lippincott Williams & Wilkins; 2001.)
1. Genetics
a. Autosomal dominant
b. FGFR1, FGFR2, FGFR3 mutations
2. Craniofacial features: Turribrachycephaly, coronal and/or sagittal synostosis, shallow orbits, hypertelorism, downslanting palpebral fissures, and midface hypoplasia
3. *Extremities: Broad thumbs and great toes, partial syndactyly of digits 2 and 3
4. Mental status: Variable
E. Jackson-Weiss syndrome
1. Genetics
a) Autosomal dominant
b) FGFR2 mutation
2. Craniofacial features: Highly variable, may appear similar to other syndromes
3. Extremities: Broad great toes or syndactyly of toes
F. Carpenter syndrome
1. Genetics: Autosomal recessive (most syndromic craniosynostosis are AD)
2. Craniofacial features: Variable suture synostosis, flat nasal bridge, low set ears, abnormal globe, and canthi
3. Extremities: Brachydactyly, syndactyly of hands and feet, and short stature
4. Mental status: Impaired
G. Boston-type craniosynostosis
1. Genetics
a. Autosomal dominant
b. MSX2 mutation
2. Craniofacial features: Craniosynostosis, soft palate cleft
3. Extremities: Short first metatarsal head, triphalangeal thumb
VI. FUNCTIONAL SEQUELAE OF CRANIOSYNOSTOSIS
A. Central nervous system
1. Varying degrees of cognitive impairment
2. Possible elevations in ICP (neurosurgical consultation is requisite in all patients)
B. Ocular
1. Exorbitism may lead to exposure keratitis and visual compromise
2. Strabismus
3. Bony orbit and ocular abnormalities may lead to deprivation amblyopia
C. Airway
1. Midface hypoplasia may result in varying degrees of airway compromise: From obstructive sleep apnea to critical airway stenosis
2. Tracheostomy may be required
3. Monobloc or Le Fort III advancement may also be required
D. Abnormal speech and hearing
VII. TREATMENT
A. Multidisciplinary team includes plastic surgeon, neurosurgeon, otolaryngologist, pediatrician, oral surgeon, orthodontist, pediatric dentist, ophthalmologist, geneticist, child neuropsychologist, speech therapist, social worker, dietician, and nurses
B. Preoperative considerations
1. Parents should be engaged in operative plan of care
2. Preoperative hematocrit with type and cross (~80% perioperative transfusion requirement)
3. Two large-bore peripheral intravenous lines, urinary catheter, and arterial line
4. ICU bed for 24 to 48 hours postoperatively
5. Perioperative antibiotics
6. Perioperative steroids may be used to decrease swelling
7. Prone versus supine positioning depending on involved suture(s) and surgeon preference
a. Special head rest may be necessary
b. Modified prone positioning
i. Greater exposure
ii. Preoperative cervical spine films required to rule out craniovertebral abnormalities
8. Ophthalmic ointment and corneal shields
9. Anesthetic considerations
a. Warming device to maintain normothermia
b. Hypotensive anesthesia
c. Cell saver devices for directed autotransfusion
d. Anti-fibrinolytics (Amicar) and erythropoietin have been used to mitigate bleeding risk
C. Operative interventions
1. Timing: 3 months to 1 year of age (earlier if evidence of elevated ICP)
2. Earlier operation: Patient maintains capacity for dural-induced ossification of small cranial defects
D. Postoperative care
1. ICU monitoring for 24 to 48 hours
2. Regular neurologic checks every 1 to 2 hours
3. Serial hematocrits to evaluate ongoing bleeding
4. Electrolyte abnormalities (especially sodium) due to disruption of hypothalamic-pituitary axis
a. SIADH (syndrome of inappropriate antidiuretic hormone): Low serum sodium, treat with fluid restriction, salt tabs, or increased sodium in IV fluids
b. Diabetes insipidus: High serum sodium and increased urine output. Treat with fluid resuscitation due to the risk of dehydration.
5. ICP monitoring (only in select cases)
6. Postoperative fever is very common; postoperative infection is rare
7. Other complications: Venous air embolism, dural lacerations, CSF leak, visual changes, seizures, meningitis, and death
VIII. CRANIOFACIAL (TESSIER) CLEFTS
A. Etiology
1. Lack of fusion of facial processes
2. Lack of migration of mesoderm
3. Possible amniotic banding
B. Extremely rare
1. Incidence: 1:100,000 births
2. *Tessier classification (Fig. 27-4)
3. “Oculocentric”
a. Cranial clefts extend superiorly from the lid margin
b. Facial clefts extend inferiorly from the lid margin
c. Corresponding cranial and facial clefts sum to 14 (e.g., 0 and 14, 1 and 13, and 6 and 8)
d. Tessier 7 cleft
i. Most common of all craniofacial clefts
ii. *Findings: ipsilateral microtia and macrostomia
e. Clefts 0-3: Oral-nasal, Clefts 4-6: Oral-Ocular, Clefts 7-9 Lateral facial
C. Soft-tissue abnormalities predict the underlying bony clefts (e.g., irregular hairline and lid margins)
D. Clefts may involve globe (coloboma) and extraocular muscles
IX. BRANCHIAL ARCH SYNDROMES AND HEMIFACIAL MICROSOMIA
A. Heterogeneous group of syndromes involving Tessier clefts 6, 7, and 8
B. Includes Treacher–Collins–Franceschetti complex, Goldenhar syndrome, and hemifacial (craniofacial) microsomia
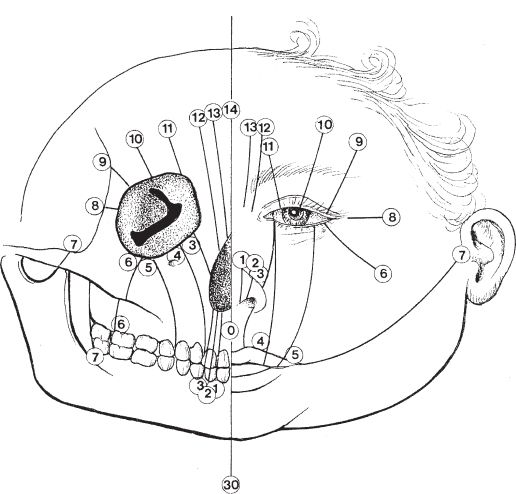
Figure 27-4. Tessier classification of craniofacial clefts.
C. Etiology
1. Vascular insults during embryogenesis (i.e., stapedial artery thrombosis)
2. Teratogens: Thalidomide and retinoic acid
3. Maternal diabetes
D. Treacher–Collins–Franceschetti complex (mandibulofacial dysostosis, Fig. 27-3C)
1. Genetics
a. Autosomal dominant
b. TCOF1 mutation (chromosome 5)
c. *Craniofacial features: Features are bilateral and symmetrical, Tessier clefts 6 to 8, with hypoplasia of body and arch of zygoma, mandibular hypoplasia, retrusion of chin, prominent facial convexity, hypoplastic lower eyelids with coloboma (congenital cleft of eyelid), absence of medial lower eyelashes, and downslanting palpebral fissures, and upper eyelids show tissue redundancy and pseudoptosis
2. Associated abnormalities
a. Microtia and middle ear anomalies result in conductive hearing loss
b. Cleft palate
c. Abnormalities of hairline
d. Airway compromise
i. Decreased pharyngeal diameter secondary to mandibular hypoplasia
ii. May necessitate early airway intervention
3. Mental status: Normal intelligence
E. Hemifacial (craniofacial) microsomia and branchial arch syndrome
1. Genetics
a. Largely sporadic, occasional familial clustering
b. Incidence: 1 in 4,000 to 5,000 live births
c. Male predominance
d. Bilateral involvement in 10% to 15% of cases
a. Variable hypoplasia of the skeleton and overlying soft tissues
b. Characteristic mandibular deformity ranging from mild hypoplasia to complete absence of the ramus, condyle, or temporal mandibular joint
c. Maxillary hypoplasia, upward occlusal cant on affected side, cross bite, open bite, and macrostomia
d. “C” deformity on frontal facial view
e. Orbital dystopia and upper lid colobomas
f. Facial muscle atrophy and weakness
g. External ear abnormalities with variable middle and inner ear anomalies
3. Mental status: Mental deficiency in 10% of cases
4. Treatment
a. Mild cases may not require treatment
b. Mandibular distraction osteogenesis during early adolescence
c. Le Fort osteotomy, bilateral split osteotomy of mandible, and genioplasty may be required in skeletally mature patients
d. External ear reconstruction
e. Audiologic evaluation and treatment for hearing loss
F. Goldenhar-Gorlin’s syndrome (oculoauriculovertebral dysplasia; Fig. 27-3D)
1. Genetics: Majority of cases are sporadic
2. Craniofacial features
a. Within the spectrum of hemifacial microsomia but more severe
b. Prominent frontal bossing, low hairline, mandibular and maxillary hypoplasia, facial muscle weakness, epibulbar dermoids (ocular dermoid tumors), preauricular skin tags and ear pits, conductive hearing loss, and vertebral abnormalities
3. Treatment: Similar to hemifacial microsomia
QUESTIONS YOU WILL BE ASKED
1. Virchow’s law.
Growth restriction occurs perpendicular to the affected suture, whereas compensatory skull growth occurs parallel to the affected suture.
2. The difference between synostotic and deformational plagiocephaly.
Deformational plagiocephaly results from external pressure applied to the pliable fetal or infant skull versus craniosynostosis which is due to premature fusion of cranial sutures and resultant compensatory growth according to properties of Virchow’s law.
3. The most common craniofacial cleft.
Tessier cleft 7 resulting in ipsilateral microtia and macrostomia.
4. Characteristic associated findings in syndromic craniosynostosis.
See Section V and Figure 27-3.
THINGS TO DRAW
1. Cranial sutures, skull bones, and pathologic head shapes in craniosynostosis
2. Tessier clefts
Recommended Readings
Bradley JP, Gabbay JS, Taub PJ, et al. Monobloc advancement by distraction osteogenesis decreases morbidity and relapse. Plast Reconstr Surg. 2006;118(7):1585–1597. PMID: 17102732.
Czerwinski M, Hopper RA, Gruss J, Fearon JA. Major morbidity and mortality rates in craniofacial surgery: an analysis of 8101 major procedures. Plast Reconstr Surg. 2010;126(1):181–186. PMID: 20220557.
Czerwinski M, Kolar JC, Fearon JA. Complex craniosynostosis. Plast Reconstr Surg. 2011;128(4): 955–961. PMID: 21681124.
Fearon JA, Ruotolo RA, Kolar JC. Single sutural craniosynostoses: surgical outcomes and long-term growth. Plast Reconstr Surg. 2009;123(2):635–642. PMID: 19182624.
Oh AK, Wong J, Ohta E, Rogers GF, Deutsch CK, Mulliken JB. Facial asymmetry in unilateral coronal synostosis: long-term results after fronto-orbital advancement. Plast Reconstr Surg. 2008;121(2):545–562. PMID: 18300974.
Smartt JM Jr, Reid RR, Singh DJ, Bartlett SP. True lambdoid craniosynostosis: long-term results of surgical and conservative therapy. Plast Reconstr Surg. 2007;120(4):993–1003. PMID: 17805129.
Tessier P, Kawamoto H, Posnick J, Raulo Y, Tulasne JF, Wolfe SA. Taking calvarial grafts, either split in situ or splitting of the parietal bone flap ex vivo—tools and techniques: V. A 9650-case experience in craniofacial and maxillofacial surgery. Plast Reconstr Surg. 2005;116(5 Suppl):54S–71S; discussion 92S–94S. PMID: 16217445.
Warren SM, Proctor MR, Bartlett SP, et al. Parameters of care for craniosynostosis: craniofacial and neurologic surgery perspectives. Plast Reconstr Surg. 2012;129(3):731–737. PMID: 22373978.
< div class='tao-gold-member'>









