ANATOMY
I. COLLAGEN: Most abundant connective tissue protein in mammals.
A. Twenty types of identified collagen; most abundant types are:
1. *Type I: Skin, tendon, and mature scar have a 4:1 ratio of type I : type III
2. Type II: Cartilage and cornea
3. Type III: Blood vessels and immature scar
4. Type IV: Basement membrane
B. Composed of high concentration of hydroxyproline and hydroxylysine amino acids.
II. SKIN LAYERS AND STRUCTURES (FIG. 1-1 A,B)
A. Epidermis: Derived from ectoderm—stratified, keratinized, and avascular layer
1. Stratum basale (aka germinativum): This layer also contains melanocytes (of neural crest origin) that produce melanin, which is taken up by the predominant keratinocytes.
2. Stratum spinosum: Desmosomes connect cells and create a shiny appearance.
3. Stratum granulosum: Cytoplasmic granules contribute to keratin formation.
4. Stratum lucidum: Dead cells without nuclei
5. Stratum corneum: Acellular layer of keratin
B. Dermis: Derived from mesoderm
1. Papillary: Loose vascular tissue
2. Reticular: Dense, more vascular layer
3. Contains fibroblasts, adipocytes, macrophages, collagen, and ground substance.
C. Adnexa: Sources of reepithelialization in partial-thickness wounds
1. Hair follicles (ectodermal origin)
a. Ingrowth of epidermis into dermis and subcutaneous tissue.
b. Associated sebaceous glands secrete into the hair follicle.
c. Retained in split-thickness skin grafts.
2. Eccrine sweat glands (ectodermal origin)
a. Coiled structures that secrete via a single duct into the epidermis.
b. Not present in split-thickness skin grafts and can lead to dryness.
3. Apocrine sweat glands (ectodermal origin)
a. In axillary and inguinal regions; secrete into hair follicles.
III. MUSCLE: Derived from paraxial mesoderm; classified as smooth, skeletal, and cardiac muscles. Muscle anatomy should be understood in this regard.
A. Microscopic: Sarcomere unit—organized myofibrils composed of actin and myosin filaments; bundles of myofibers form muscle fibers.
B. Macroscopic: Organized groups of muscle fibers for fascicles; bundles of fascicles form muscles.
C. Neuromuscular junction: “Motor end plate” consists of sarcolemmal folds within which acetylcholine receptors reside.
______________
*Denotes common in-service examination topics
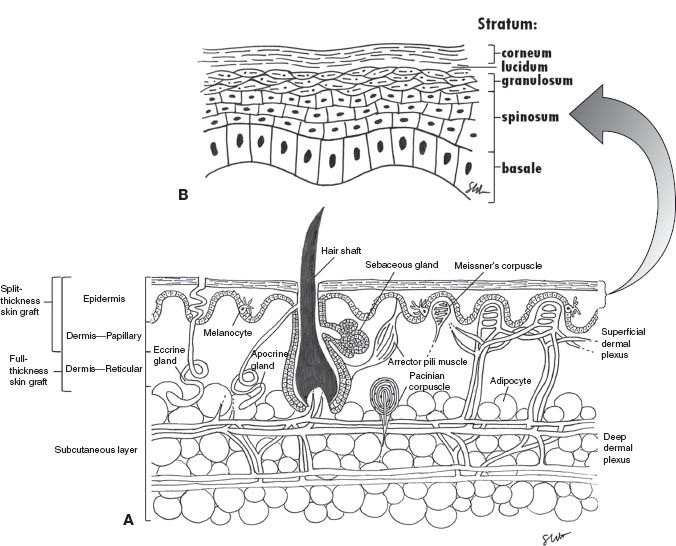
Figure 1-1. Cross section of the skin. A: All skin layers. B: Epidermis. Meissner and Pacinian corpuscles only in glaborous skin.
IV. BONE: Derived from lateral plate mesoderm (except for skull bones derived from neural crest)
A. Cross-sectional anatomy
1. Outer layer: Fibrous periosteum and osteogenic periosteum (these cells participate in fracture repair).
2. Mature compact (cortical) bone: Eighty percent of total bone mass; lamellar structure that is permeated by elaborate interconnecting vascular canals (Haversian canals).
3. Immature compact (cortical) bone: Woven structure of collagen fibrils that is replaced by mature bone through remodeling.
4. Trabecular (cancellous) bone: Only 20% of total bone mass, but much greater surface area due to lower density; bony matrix organized into a matrix (trabeculae) along lines of stress. Develops into compact bone via osteoblasts along the trabeculae.
V. TENDON: Derived from lateral plate mesoderm
A. Organizational anatomy
1. Collagen is arranged longitudinally into fibrils.
2. Fibrils and fibroblasts are organized into fascicles, which are grouped into tendons.
VI. CARTILAGE: Derived from lateral plate mesoderm, the cartilage consists of extracellular matrix (ECM) composed of collagen fibers, ground substance, and elastin, and is classified into elastic cartilage, hyaline cartilage, and fibrocartilage, depending on the proportion of each component.
A. Cross-sectional anatomy
1. Zone 1: Superficial, uncalcified cartilage
2. Zone 2: Intermediate cartilage
3. Zone 3: Deep cartilage (abutting bone)
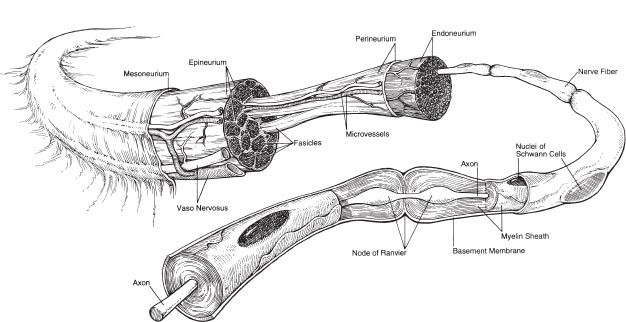
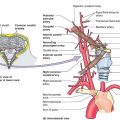
Figure 1-2. Anatomy of peripheral nerve. Nerves are composed of fascicles, each of which contain multiple nerve fibers (axons).
VII. NERVE: Peripheral nerves have neural crest origin
A. Organizational anatomy (Fig. 1-2)
1. Clusters of cell bodies or ganglia
2. A nerve describes a bundle of axons traveling together peripherally
3. The majority of axons are myelinated and individual axons are enveloped in endoneurium
4. Bundles of axons are called fascicles and are wrapped in perineurium
5. The nerve is covered by epineurium
NORMAL WOUND HEALING
VIII. SKIN AND SUBCUTANEOUS TISSUE
A. Wound healing categories
1. Primary intention
a. Immediate primary closure of a surgical incision *(epithelialization occurs in ~24 hours).
b. Delayed closure of a surgical incision (usually to either allow clearance of infection or resolution of edema) is known as “delayed primary closure”.
2. Secondary intention
a. Full-thickness wound healing by a combination of wound contraction and migration of fibroblasts and keratinocytes from the wound periphery.
B. Overview of the phases of wound healing
1. Inflammatory phase (first minutes to first week)
a. Vasoconstriction of vessels for first 10 minutes after injury.
b. Coagulation: *Platelets arrive and degranulate, releasing thromboxane A2 that causes transient vasoconstriction to facilitate hemostasis with thrombus formation. PDGF and TGFB are also released.
c. Vasodilation and increased permeability: Small vessels dilate in response to prostaglandins to allow white blood cells (neutrophils, plasma cells, and monocytes) attracted by the leukotrienes, complement, and cytokines (inter-leukin-1 [IL-1], tumor necrosis factor-α [TNF-α], transforming growth factor-β [TGF-β], and platelet factor 4 [PF4]) to enter.
d. Cellular response
i. Neutrophils
a) Dominant cell type at 24 hours
b) Approach injury site by chemoattractants via circulatory system
c) Undergo magination and diapedsis
d) Migrate through interstitium by chemotaxis to injury site
ii. Macrophages (transformed monocytes) are the dominant cell type at 2 to 3 days, releasing cytokines to attract fibroblasts
2. Proliferative phase (aka “fibroblastic phase,” ~days 3 to 14)
a. *Fibroblasts are the predominant cell population at 3 to 5 days and transform into myofibroblasts to promote wound contraction under the influence of platelet-derived growth factor (PDGF) and TGF-β1 released by macrophages.
b. High rate of collagen synthesis from days 5 to 21.
c. Tensile strength begins at days 4 to 5
d. Fibroblasts form ECM by synthesizing proteoglycan and fibronectin (which is then replaced by collagen).
e. *Keratinocytes migrate into the wound starting with the loss of contact inhibition.
f. *Neovascularization occurs under the influence of vascular endothelial growth factor expression.
3. Remodeling (maturation) phase (~week 3 to 1 year)
a. Collagen replaces proteoglycan/fibronectin and reorganizes creating stronger crosslinks.
b. Equilibrium between collagen breakdown and synthesis by weeks 3 to 5.
c. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) remodel the collagen matrix.
d. The wound achieves 3% of its original strength at 1 week, 30% original strength at 3 weeks, and 80% original strength at 2 months and beyond. *Final ratio of type I:type III collagen is 3.5:1.
C. Epithelialization
1. Mobilization: Loss of contact inhibition.
2. Migration: Cells migrate across the wound until meeting cells when the contact inhibition sets in.
3. Mitosis: Cells further back from wound edge proliferate to bridge wound.
4. Differentiation: Reestablishment of epithelial layers from basal layer to stratum corneum after migration ceases.
D. Contraction (happens when full-thickness injury through dermis is present). Fibroblasts transform into myofibroblasts
1. Myofibroblasts are present throughout granulating wound
2. Myofibroblasts appear at day 3 and reach the maximum level at days 10 to 21.
3. Less contraction when more dermis within the wound
IX. PHASES OF MUSCLE HEALING
A. Phases of muscle healing (phases overlap with each other)
1. Destructive phase (days 0 to 7 following injury). Myoblasts join with each other to form myotubes which then fuse to form new myofibers.
a. Analogous to inflammatory phase of skin healing
b. Inflammatory response with cytokine release
c. Initial neutrophil response followed by macrophages
2. Repair phase (starting at day 3, lasting up to several weeks)
a. Regeneration of disrupted myofibers
b. Production of connective tissue scar
3. Remodeling phase (occurs concomitantly with repair phase)
a. Vascular ingrowth (to feed the upregulated metabolism of regeneration)
b. Regeneration of intramuscular nerves is necessary for functional regeneration
c. Adhesion of myofibers to ECM
X. BONE
A. Bone healing categories
1. Primary (direct) bone healing by surgical fixation
a. Minimal callus formation (bypasses the stage of woven bone formation)
b. Lamellar bone formation parallel to the long axis of the bone
2. Secondary (indirect) bone healing by external splint/cast fixation
a. Typical callus formation, amount of callus correlates with the amount of instability encountered during healing.
b. Immobilization is important to allow for healing.
1. Inflammatory phase (from time of fracture and tapering off at when bone formation starts at 7 to 10 days)
a. Initial platelet degranulation and contained hematoma aids in healing.
b. Inflammatory response as detailed in previous section; osteoclasts break down necrotic bone edges, releasing osteogenic cytokines.
2. Reparative phase (starting during the first week and lasting up to several months)
a. Inflammatory debris is cleared by macrophages.
b. Acid tide—acidic local environment stimulates osteoclasts.
c. Vascular ingrowth from periosteum and endosteum.
d. pH rises at ~day 10 with the presence of increased alkaline phosphatase, leading to the formation of newly woven bone at the edges.
e. At ~3 weeks callus fills in between the edges (starts a soft callus populated by chondrocytes, which gradually calcifies into hard callus by endochondral ossification); continued bone formation by osteoblasts leads to bony edge unification.
3. Remodeling phase (starting after fracture solidly united at 2 to 3 months and continuing for years)
a. Woven bone is slowly replaced by the lamellar bone according to the Wolff law; medullary canal is restored.
b. “Clinical healing” (defined as the state of adequate stability and resolution of pain to allow protected motion) occurs in most bones by 4 to 6 weeks. Radiographic healing may lag by 6 months.
C. Bone grafting
1. Aspects of healing: An ideal bone graft (autogenous, cancellous) possesses osteoconductive, osteoinductive, and osteogenetic properties
a. *Osteoconduction—donor bone placed adjacent to the recipient bone will allow ingrowth of capillaries and osteoprogenitor cells, ultimately resulting in complete incorporation. Acts as a scaffold.
b. *Osteoinduction—active induction of differentiation of osteoblast precursors into bone-forming cells. Bone morphogenetic proteins: 2, 4, and 7 isotypes have the most significant osteoinductive effects.
c. *Osteogenesis—formation of new bone by virtue of osteogenic precursors present within a graft.
XI. TENDON
A. Two mechanisms of tendon healing categories
1. Intrinsic healing
a. Tendon’s intrinsic capacity to heal (operative repair aims to maximize this type of healing)
b. Mediated by tenocyte/fibroblast population that arises from the tendon and epitenon.
c. Relies on synovial diffusion for nutrition
d. Enhanced by mobilization
2. Extrinsic healing
a. Surrounding soft tissue’s tendency to repair damaged tendon
b. Ingrowth of inflammatory cells and fibroblasts overlying the sheath
c. Immobilization leads to the formation of debilitating adhesions to tendon, limiting range of motion (early mobilization minimizes adhesions caused by extrinsic healing).
B. Phases of healing
1. Inflammatory phase (within first few days, inflammatory response peaking at 3 days)
a. Tendon defect fills with hematoma, tissue debris, and fluid
b. Both intrinsic cells and cells that have migrated from the periphery bridge the defect
c. Increased phagocytic activity clears necrotic debris
2. Proliferative phase (starting at ~day 5 and lasting up to several weeks)
a. Fibroblasts are the predominant cell type, proliferating from epitenon and endotenon.
b. Collagen initially deposited perpendicular to the tendon axis; at ~4 weeks, the collagen fibers realign to the long axis.
c. Strength of repair begins and increases at ~2 to 3 weeks; synovial sheath is reconstituted at 3 weeks.
d. Vascular ingrowth occurs
3. Remodeling phase (starting at several weeks after injury and lasting up to 1 year after):
a. Collagen fibers continue to realign to the long axis of the tendon
b. Fibers realigned by 8 weeks
C. Tendon grafts
1. Extrasynovial tendon grafts (most commonly include palmaris longus or plantaris)
a. Early cell death and eventual repopulation by fibroblasts and neovascularization.
b. Acts as a conduit for vessel and cell ingrowth
2. Intrasynovial tendon grafts (rare; e.g., Flexor digitorum longus)
a. Cellular viability is thought to be maintained, and healing occurs by the normal mechanisms.
b. Less adhesion formation
XII. CARTILAGE
A. Avascular tissue without intrinsic healing potential
B. Healing initiated by damage to the surrounding tissue (e.g., perichondrium and subchondral bone)
C. Extra-articular cartilage versus intra-articular cartilage healing
1. Extra-articular cartilage (e.g., auricular and nasal) injury
a. Tissue injury response generated by perichondrium with fibroblast influx and scar formation (but not true regeneration of cartilage).
2. Intra-articular cartilage injury
a. Superficial (without violation of subchondral bone)—no blood-carrying progenitor cells are released, thus no repair occurs.
b. Full-thickness (through cartilage and into subchondral bone)—allows influx of progenitor cells and formation of fibrocartilage. Fibrocartilage is less organized, more vascular, less tolerant of mechanical force, and more susceptible to degradation compared with normal cartilage. Fibrocartilage eventually breaks down, resulting in an arthritic joint.
XIII. NERVE
A. Response to injury
1. Trauma to vasa nervorum and surrounding tissue leads to inflammatory response.
2. If the injury is close to the neuron cell body, the entire neuron may die (e.g., brachial plexus avulsion injuries).
3. Typical injuries to nerves in peripheral locations (e.g., complex forearm laceration) will affect connective tissues (Schwann cells) and the axon but not the actual neuronal cell body.
4. Wallerian degeneration: Schwann cells will die, and the distal axon degrades. This can extend up to 2 cm proximal to the injury site.
5. Axon degradation and clearing of debris takes 15 to 30 days and precedes nerve regeneration.
6. Axonal regrowth occurs in response to neurotrophins (e.g., brain-derived neurotrophic factors, ciliary neurotrophic factor, and nerve growth factor) secreted by target cells (postsynaptic neurons or muscle cells) and by Schwann cells.
7. Macrophages secrete interleukins that induce Schwann cell proliferation.
8. Schwann cells along the distal axonal tract express laminins and adhesion molecules, which help guide the regenerating axon.
9. Axonal sprouts from the proximal cut end must enter the distal tract to regrow. If disruption of the nerve is severe and/or scarring is great, the budding axons cannot cross the gap, and regeneration does not occur.
10. Muscles innervated by the injured nerve will atrophy (70% loss at 2 months). Some muscle fibers die at 6 to 12 months if there is no regeneration of nerve. Motor end plates remain open for approximately 1 year (variable) before fibrosis develops, making reinnervation of that particular muscle impossible.
11. *Once growth is initiated, axons extend by approximately 1 mm a day
B. *Seddon nerve injury classification
1. Neuropraxia
a. Segmental interruption of myelin sheath leading to local transient block of conduction along a nerve.
b. The anatomy of the nerve is preserved, and no Wallerian degeneration occurs.
c. Recovery is usually rapid (few weeks), but may take several months.
d. Selective demyelination of fibers may occur.
2. Axonotmesis
a. Axonal damage within the nerve. Schwann cell basal lamina (inner endoneurial sheath) is preserved.
b. Wallerian degeneration occurs. Recovery rate is 1 mm/day along the nerve, once healing begins.
c. Fibrillations are present on electromotor testing.
d. Recovery is typically complete eventually (without surgery) if axonal regeneration is able to progress across the injury zone.
3. Neurotmesis
a. Nerve is transected with destruction of nerve, myelin sheath, and surrounding connective tissue; Wallerian degeneration occurs.
b. Spontaneous recovery does not occur; surgical repair is needed for the best outcome.
C. Sunderland/Mackinnon nerve injury classification (Table 1-1)
1. First-degree injury
a. Nerve is demyelinated, resulting in a local conduction block.
b. Treatment is nonoperative and recovery is complete within approximately 12 weeks.
2. Second-degree injury
a. Some nerve fibers are disrupted, but the Schwann cell basal lamina remains intact.
b. Wallerian degeneration occurs with second-degree and higher injuries.
c. Tinel’s sign indicates an advancing growth cone.
d. Treatment is nonoperative. Complete recovery is expected in months.
3. Third-degree injury
a. Some areas of Schwann cell basal lamina are disrupted with scarring, while the perineurium remains intact.
b. Incomplete recovery; some nerve fibers do not reinnervate their target.
c. Treatment is usually nonoperative.
4. Fourth-degree injury
a. Loss of continuity of the perineurium.
b. Scar blocks all fiber growth; little or no nerve recovery.
c. Treatment is operative.
5. Fifth-degree injury
a. The nerve is completely transected.
b. The epineurium is disrupted.
c. No recovery is expected without operative management.
6. Sixth-degree injury
a. Combination of any of the previous five levels of injury.
PATHOLOGIC WOUND HEALING
I. WOUND FAILURE (SKIN, SUBCUTANEOUS TISSUE, FASCIA, MUSCLE)
A. Acute wound failure (dehiscence): Postoperative separation of the surgical incision
1. Occurs when the load applied to the wound exceeds the strength of the suture line and provisional matrix.
2. Most commonly happens at 7 to 10 days postoperatively, can happen any time from day 1 to more than 20 days after surgery.
a. Surgeon factors
i. Technical error (most common cause for early fascial dehiscence within several days after abdominal surgery)
ii. Emergency surgery
b. Systemic factors
i. Advanced age
ii. Chronic corticosteroid therapy
iii. Malnutrition
iv. Radiation therapy
v. Chemotherapy
vi. Systemic disease (jaundice, renal failure, and diabetes)
c. Local factors
i. Hematoma
ii. *Seroma (the most common cause of late postoperative skin dehiscence following post-bariatric body contouring)
iii. Infection
iv. Edema
v. Excessive tension (noncompliance with following activity restrictions)
vi. Elevated intra-abdominal pressure (for abdominal closures)
vii. Previous wound dehiscence
B. Chronic wound failure (nonhealing wounds)
1. Failure to achieve anatomic/functional integrity over 3 months
2. Diabetes, venous stasis, ischemic tissue loss, and pressure sore are common etiologies
3. Underlying osteomyelitis, hidradenitis, or pyoderma gangrenosum
4. Possibility exists for the development of squamous cell carcinoma (aka Marjolin’s ulcer) in the setting of a chronic wound
5. Associated physiologic derangements
a. Cytokine abnormalities: Increased IL-1, IL-6, TNF-α; decreased epidermal growth factor, PDGF.
b. *Abnormal ECM dynamics: Increased MMPs, decreased TIMPs
6. Associated factors
a. Local factors
i. Infection
ii. Infected foreign body (e.g., orthopedic hardware)
iii. Ischemia (arterial insufficiency or pressure-related)
iv. Venous insufficiency (leads to protein extravasation, edema, and decreased oxygen diffusion)
v. *Radiation therapy leads to vascular fibrosis (relative ischemia) and decreases mitotic potential of fibroblasts (also consider possibility of osteoradionecrosis of the bone).
b. Systemic factors
i. Hypoxia
ii. Smoking
iii. Diabetes (microvascular and macrovascular diseases leading to local ischemia; glycosylation of hemoglobin impairs oxygen delivery; impaired neutrophil function; peripheral neuropathy)
iv. Chronic disease
v. Advanced age (shortened inflammatory phase causing decreased strength of healing)
vi. Malnutrition
a) Vitamin C
1) *Collagen cross-linking by hydroxylation of proline and lysine
2) *Lack of vitamin C leads to “scurvy”: Low collagen tensile strength manifests in collagen-containing tissues (skin, dentition, bone, and blood vessels) as hemorrhage (petechiae and swollen gums), loss of dentition, and impaired bone healing.
b) *Folate and vitamin B6 (pyridoxine): DNA synthesis and cellular proliferation
c) *Vitamin E: Strong antioxidant and immune modulator
d) *Zinc: Cofactor for numerous metalloenzymes and proteins; necessary for protein and nucleic acid synthesis.
e) *Assess nutrition with albumin level (normal >3.5 g/dL, 20 day half-life) or pre-albumin level (normal >17 g/dL, 3 day half-life)
vii. Chemotherapy: Most detrimental agents are doxorubicin, cyclophosphamide, methotrexate, bischloroethylnitrosourea (BCNU), and nitrogen mustard
viii. Glucocorticoids
a) Inhibit the inflammatory phase and inhibit collagen synthesis of fibroblasts, leading to decreased wound strength.
b) *Can reverse effect with oral vitamin A to augment epithelialization and fibroblast proliferation
ix. Tamoxifen (dose-dependent effect)
x. *Anemia by itself does not impair wound healing
II. BONE—PATHOLOGIC HEALING
A. Types of bone healing pathology
1. Delayed union: When clinical healing is delayed beyond the usual expected time with radiographic evidence of inadequate osteocyte activity and deficient callus formation.
2. Nonunion: When there is no evidence of clinical or radiographic healing beyond the usual healing time, often with a mobile area fibrous scar and interposed tissue in the gap (pseudoarthrosis).
a. Atrophic nonunion: Marked resorption of the bony ends at the fracture site without callus (therapeutic intervention may involve provision of internal fixation along with an osteoinductive stimulus such as bone graft).
b. Hypertrophic nonunion: Significant callus formation, but no bridging of fracture (usually result of failure to provide stable fixation).
B. Factors detrimental to bone healing
1. Local factors: Soft tissue crush/loss, soft tissue interposition into fracture gap, open fractures, segmental fractures, articular fractures, infection, pathologic fractures, extensive soft tissue stripping, inadequate reduction, inadequate immobilization, rigid fixation with gap, distraction of fracture, and delay in treatment
2. Systemic factors: Anemia, malnutrition, vitamin D deficiency, growth hormone deficiency, diabetes, smoking, NSAIDs, steroids, and anticoagulants
III. TENDON—PATHOLOGIC HEALING
A. Immobilization after primary tendon repair
1. Extrinsic healing predominates with tendon sheath adhesion formation.
2. Disorganized collagen fibrils and decreased strength of repair.
B. Overuse tendinosis: Painful condition beginning with repetitive microtrauma to tendon; characterized by degenerative changes in tendon.
1. Aging, inflammatory cytokines, and ischemia are thought to be contributing factors
2. May be seen with or without inflammation of paratenon
C. Postoperative rupture of tendon repairs
1. Attributable to a greater load encountered than the strength of repair (repair is weakest between days 6 and 18, peak occurrence at day 10).
2. Poor tendon healing due to gapping caused by poor surgical technique
3. *Fluoroquinolones inhibit tenocyte metabolism, reducing cell proliferation and collagen/matrix synthesis and can lead to spontaneous rupture of tendons.
IV. NERVE—PATHOLOGIC HEALING
A. Neuroma—painful regrowth of nerve in a scarred area of previous injury.
B. Failure of axonal regeneration (potential for axonal regeneration decreases with age)
1. Degeneration of sensory receptors (for sensory nerves)
2. Fibrosis of motor end plates (for motor nerves)
C. Cross-innervation (e.g., facial synkinesis, gustatory sweating [Frey’s syndrome])
NORMAL SCARRING
I. VISIBLE SCAR IS THE NORMAL ENDPOINT FOR ALL FULL-THICKNESS SKIN INJURIES
A. Factors that lead to less conspicuous scars
1. Older age
2. Lighter colored skin
3. Surgical incision as opposed to traumatic laceration
4. Placement of incision or laceration within (parallel to) relaxed skin tension line
5. Minimal tension following closure (e.g., eyelids)
6. Optimal surgical technique (e.g., atraumatic manipulation, skin edge eversion, and removal of suture in 5 to 7 days on face)
PATHOLOGIC SCARRING
I. HYPERTROPHIC SCAR
A. Definition: An abnormal wound healing endpoint in response to trauma, inflammation, burn, or surgery
1. Raised, erythematous, and often pruritic
2. *Remains within the boundaries of original wound
3. Upregulated fibrogenic cytokines (TGF-β isoforms, PDGF, and insulin-like growth factor 1 [IGF-1]) lead to higher levels of collagen synthesis
B. Etiology
1. Major factors
a. Amount and depth of trauma (most commonly with burns)
b. Inflammation, infection
c. Prolonged open wound (>21 days, most commonly with burns)
2. Contributing factors
a. Areas of tension
b. Darker skin tone
C. Natural history
1. Becomes apparent at ~6 to 8 weeks after injury
2. Worsens over 6 months
3. May cause contractures at joints
4. May take 1 to 2 years to mature (scar will become less red, less tender, and less pruritic)
5. May regress somewhat without any intervention at all
D. Histologic characteristics (under standard light microscopy, hypertrophic scar and keloid are indistinguishable)
1. Cigar-shaped nodules of blood vessels, fibroblasts, and collagen fibers that are arranged parallel to epidermis and oriented along tension lines (normal skin: the basket-like woven pattern of collagen fibers)
2. *Presence of α-smooth muscle actin producing myofibroblasts (not present in keloids)
3. Lower ratio of type I:type III collagen (2:1). (Type I:type III collagen ratio is 3.5:1 in normal scars.)
E. Treatment approach
1. Nonoperative
a. Pressure garments
i. Commonly used for hypertrophic burn scars
ii. Induces local tissue hypoxia, reduces fibroblast proliferation and collagen synthesis
iii. Compression of 24 to 30 mmHg to be effective
b. *Silicone sheeting and topical silicone gel
i. Unclear mechanism of action—thought to increase hydration of remodeling scar
ii. Require application of at least 12 hours/day for at least 3 months to be effective
c. Corticosteroid injection
a. Attention to atraumatic technique, excision of inflamed tissue, avoidance of nidus for inflammation (e.g., trapped hair or unnecessary deep resorbable suture), and tension-free closure.
b. Z-plasty tissue rearrangements to release contractures
c. May require graft or flap reconstruction for coverage
d. Fractional ablative CO2 laser can be helpful adjunct
II. KELOIDS
A. Definition: An abnormal wound healing endpoint in response to trauma, inflammation, burns, or surgery.
1. May start as a raised, erythematous, and pruritic lesion
2. *Evolves into an enlarging mass that extends beyond the original boundaries of the wound.
3. Higher level of collagen synthesis compared with hypertrophic scars due to upregulated fibrogenic cytokines (TGF-β isoforms, PDGF, and IGF-1) and increased number of receptors for these cytokines within keloidal fibroblasts.
4. *Increased fibroblast proliferation
5. *Absence of myofibroblasts and decreased density of blood vessels in comparison to hypertrophic scars.
6. Decreased expression of MMPs (that degrade ECM)
7. Increased levels of adenosine triphosphate within keloid
B. Etiology
1. Major factors
a. Darker skin tone
b. Genetic predisposition
2. Contributing factors
a. Age (peak just after puberty)
b. Hormones (keloids worsen during puberty and pregnancy; postmenopausal women experience softening and flattening of keloids)
C. Natural history: Evolves over time without a significant regression or quiescent phase
D. Histologic characteristics (under light microscopy, hypertrophic scar and keloid are indistinguishable)
1. Thick and large collagen fibers haphazardly packed closely together
2. *Much higher ratio of type I:type III collagen (18:1)
E. Treatment approach: Nonoperative and operative interventions are required, and an extremely high rate of recurrence persists (50% to 80%)
1. Nonoperative
a. Pressure devices (e.g., pressure clip for earlobe)
b. Silicone sheeting and topical silicone gel
c. Corticosteroid injection
d. Radiation therapy
2. Surgical
a. Attention to atraumatic technique, excision of inflamed tissue, avoidance of nidus for inflammation (e.g., trapped hair or unnecessary deep resorbable suture), and tension-free closure
b. Excision ± skin graft depending on the size of the lesion
III. COLLAGEN SYNTHESIS DISORDERS
A. Ehlers–Danlos syndrome
1. Autosomal dominant transmission
2. Significant joint laxity, thin and friable skin, and severe skin hyperlaxity
3. Extreme risk for wound healing problems (e.g., recurrent ventral hernia)
4. Increased risk of infection (inherently defective immune response)
5. Should be discouraged from pursuing elective and aesthetic surgery
1. Defective collagen synthesis results in hyperextensible skin
2. Can be autosomal dominant, autosomal recessive, or X-linked
3. Autosomal and X-linked variants have more generalized manifestations: Growth retardation, skeletal dysplasia, facial dysmorphia, emphysema, cardiovascular involvement, hernias, and hollow viscus diverticula
4. *Not a contraindication to surgery
PEARLS
1. Scars typically widen over time. Some areas, such as the back or the legs, are especially prone to scar widening.
2. Nicotine in any form (smoking, patches) impairs wound healing significantly due to vasoconstrictive effects
3. Macrophages are critical cells in wound healing and initiate the growth factor cascade, fibroblast proliferation, and collagen formation
4. Prior to considering scar revision, at least 1 year should pass to allow for complete scar remodeling
QUESTIONS YOU WILL BE ASKED
1. What is the difference between wound contraction and wound contracture?
Wound contraction is a part of secondary healing beginning a few days after injury as myofibroblasts contract and reduce the size of the wound to be epithelialized. Wound contractures occur when bands of collagen are deposited at the site of hypertrophic scar formation; these are termed “contractures” when they impair functionality (e.g., hands) or range of motion (e.g., axillae and neck).
2. What is the difference between hypertrophic scar and keloid?
Hypertrophic scar does not extend beyond the borders of the original wound, whereas keloids grow well beyond these borders; histologically, these two fibroproliferative disorders are different, but they are indistinguishable under standard H&E preparation on light microscopy. They have much different type I:type III collagen ratios. Hypertrophic scars produce smooth muscle actin by myofibroblasts, whereas keloids do not.
3. What are the factors that impair wound healing?
Systemic conditions (e.g., diabetes, autoimmune conditions, and medications), ischemia, pressure injury, infection, malignancy, foreign body, venous insufficiency, irradiation, hypoxia, smoking, advanced age, and malnutrition.
4. What are the types of nerve injury and their expected recovery? Which types require surgical intervention?
See “NORMAL WOUND HEALING” → “NERVE” → Sections B and C.
Recommended Readings
Broughton G, Janis JE, Attinger CE. The basic science of wound healing. Plast Reconstr Surg. 2006;117(7 Suppl):12S–34S. PMID:16799372.
Garner WL, Rahban SR. Fibroproliferative scars. Clin Plast Surg. 2003;30(1):77–89. PMID: 12636218.
Maggi SP, Lowe JB 3rd, Mackinnon SE. Pathophysiology of nerve injury. Clin Plast Surg. 2003;30(2): 109–126. PMID: 12737347.
< div class='tao-gold-member'>