History
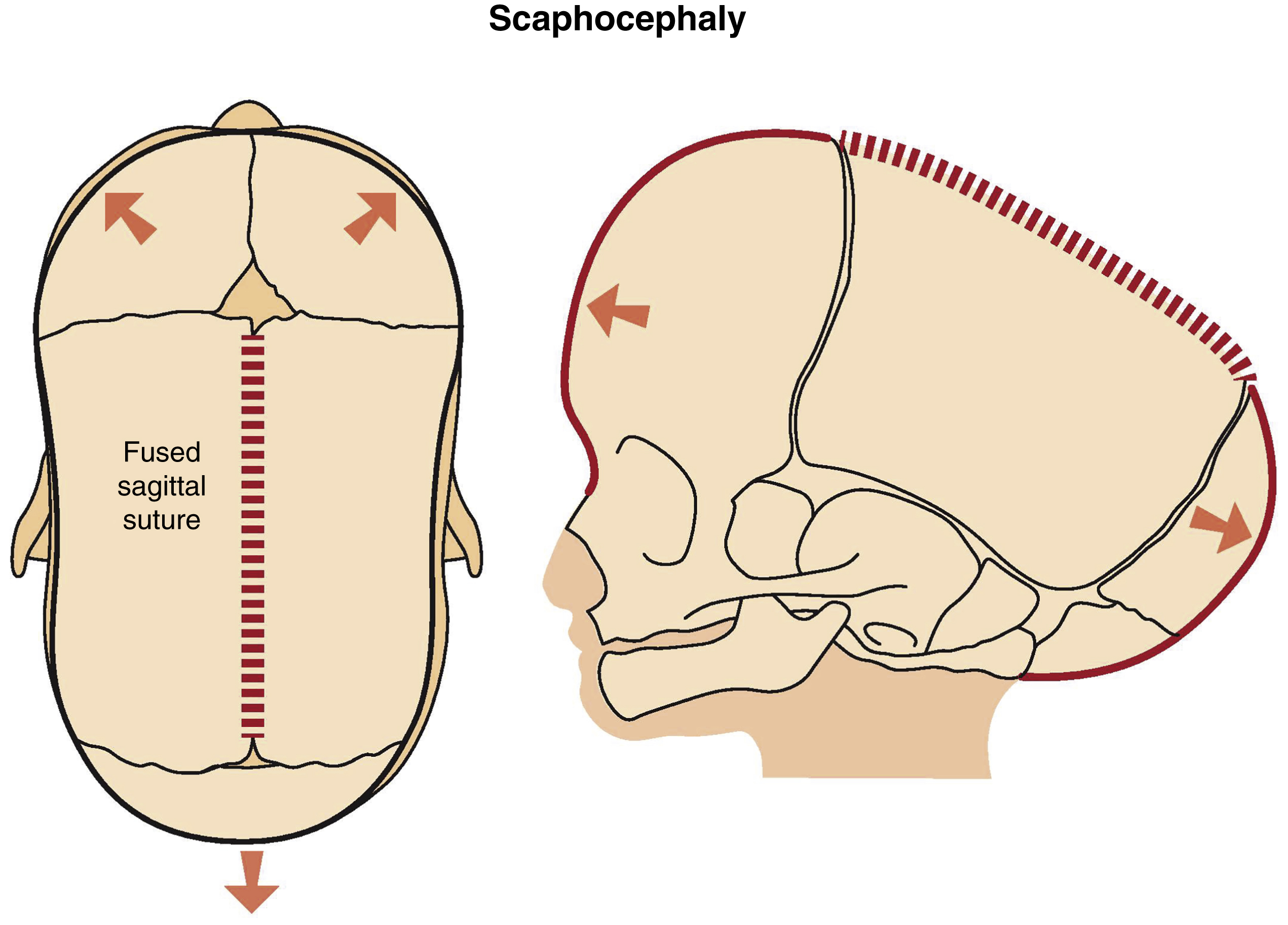
Hippocrates provided the first description of what is now called craniosynostosis in 100 BC and noted the variation of skull deformities and correlated it with the pattern of cranial suture involvement. There are many historical descriptions of people with unusual head shapes. The Greek statesman and General Pericles had an unusually long, narrow head shape and was nicknamed “Squill head,” and he might have had sagittal synostosis. Aristophanes and Galen both used the term oxycephalus for “tower head.” The term craniosynostosis was first used by Otto in 1830 to describe the entity of premature suture fusion. Virchow described how in medieval Saxony, people believed that such children were thought to be changelings or monsters and that it was the devil who made them deaf, dumb, and blind. Virchow examined skulls from various museums and published a classification where he correlated synostosis of specific cranial sutures with head shape. What has become known as Virchow’s law is that premature fusion of a cranial vault suture (craniosynostosis) inhibits skull growth perpendicular to the fused suture and compensatory overexpansion takes place at patent sutures. The general direction of growth after synostosis is parallel to the fused suture. For example, when synostosis of the sagittal suture occurs, growth proceeds in an anteroposterior direction, leading to scaphocephaly and a long, narrow, boat-shaped skull ( Fig. 20.1 ).
Skeletal Development
The developing skull is divided into the neurocranium, which surrounds the brain, and the viscerocranium, which develops into the facial bones. The neurocranium is made up of bones that ossify directly, the membranous neurocranium, which forms the skull vault, and bones that undergo endochondral ossification and form the skull base. The flat bones of the skull grow by the deposition of new bone material at their margins. In their growth phase, these bones do not fuse but remain separated by specialized structures, the cranial sutures. Skull growth is needed to accommodate the size of the growing brain and depends on an exchange of signals between mesenchyme, the osteogenic front, and the dura mater.
Normal Calvarial Growth
Normal growth of the calvarium is complete at about age 2 years, the orbit at age 6 years, and the rest of the facial skeleton at about age 16–17 years. Brain growth drives the enlargement of the skull in infancy. The expanding brain doubles in size in the first 6 months of life and then doubles again in the second 6 months and doubles again between 12 and 24 months. The expanding brain creates separating tensional forces in the cranial sutures. The cranial sutures allow for skull deformation during birth and expansion during brain growth. The metopic suture normally starts to close at the age of 2 years. The sagittal suture begins to close after the age of 22 years, the coronal suture after 24 years, and the lambdoid after 26 years. It is within the normal range for them not to fully close until the age of 40 years.
Craniosynostosis
The prevalence of craniosynostosis ranges from 3.1 to 7.2 in 10,000 live births. Fusion can occur in one or in multiple sutures and in 21% of the cases fusion is caused by a known gene mutation. , . In nonsyndromic craniosynostosis one suture is fused without additional birth deficits and can affect any cranial suture. Sagittal synostosis is the commonest, with a prevalence of 40%–60%. Overall, the incidence of craniosynostosis has increased, but especially a rise of metopic synostosis has been reported (25% of nonsyndromic craniosynostosis), currently the second most common type, followed by unicoronal synostosis (<15%) – an unexplained change that occurred in Europe and the United States. , The rarest type is lambdoid synostosis (1% of all craniosynostosis and 3% of nonsyndromic craniosynostosis). Craniosynostosis usually occurs as an isolated condition, but has been reported to be associated with more than 200 syndromes. In syndromic craniosynostosis one or multiple sutures are fused with the presence of other congenital anomalies. Associated phenotypical features can include calvarial and facial hypoplasia and dysmorphology, hydrocephalus, deafness, blindness, mental retardation, and extremity anomalies. Craniosynostosis can have functional consequences.
Classification of Craniosynostosis
Primary
- •
Nonsyndromic: sagittal, metopic, coronal, multisuture, lambdoid, frontosphenoidal
- •
Syndromic: e.g., Muenke, Crouzon, Pfeiffer, Apert, Saethre–Chotzen.
Secondary
- •
Metabolic disorder (e.g., hyperthyroidism, hypercalcemia, hypophosphatasia, sickle cell disease and thalassemia, vitamin D deficiency, renal osteodystrophy)
- •
Malformations (e.g., holoprosencephaly, microcephaly, shunted hydrocephalus, encephalocele)
- •
Exposure of fetus to teratogenic drugs (e.g., sodium valproate, phenytoin, retinoic acid)
- •
Mucopolysaccharidosis (e.g., Hurler syndrome, Morquio syndrome).
Molecular Mechanisms Involved in Craniosynostosis
Genes and Clinical Presentation: Nonsyndromic Craniosynostosis
Nonsyndromic craniosynostoses, capturing 60% of all craniosynostosis, are mainly sporadic, however due to advances in genetic diagnostics, nonsyndromic single suture patients are increasingly recognized as syndromic patients. An example of this is the discovery of the FGFR3 P250R mutation in patients with (bi)coronal synostosis resulting in Muenke syndrome. Moreover, an increasing incidence among first-degree relatives (2%–10% of the patients) is seen, mostly in patients with metopic synostosis followed by multi-, sagittal, lambdoid and coronal suture. Sagittal synostosis is thought to be the most isolated. Multisuture synostosis without known genetic cause has the highest frequency of associated symptoms, such as ear infections, palatal abnormalities, and hearing problems.
The genetic causes of nonsyndromic craniosynostosis remain largely unknown. Mutations in the genes causing syndromic craniosynostosis and chromosomal abnormalities are not a common cause of nonsyndromic craniosynostosis. Nonsyndromic craniosynostosis as a ‘developmental pathway disorder’ has been suggested. On the latter, a stronger influence of genetic factors in the etiopathogenesis of nonsyndromic craniosynostosis has been implied. The SMAD6 mutation was found in 7% of patients with midline (sagittal and/or metopic) nonsyndromic craniosynostosis and in 25% of the patients with familial occurrence of midline craniosynostosis. Moreover, mutations such as ALX4 , BBS9 , BMP2 , EFNA4 , IGF1R , RUNX2 , SMURF1 , SPRY1 , SPRY4 are thought to be responsible for the fusion of a specific single suture.
Genes and Clinical Presentation: Syndromic Craniosynostosis
The best understood skull developmental cascades include bone morphogenetic proteins (BMP)/transforming growth factor beta (TGFβ), fibroblast growth factor (FGF), hedgehog (HH), eph-ephrin and wingless-type MMTV integration site family (WNT) signaling pathways. Most of these pathways are known to play a role in neural crest development, hence driving the morphogenesis of the craniofacial complex. There is good evidence that the primary pathology in craniosynostosis is within the dura mater and not within the bone. Cranial suture fusion and maintenance of patency are dependent on the interplay of transcription factors, cytokines, growth factor receptors, and extracellular matrix molecules.
The majority of craniosynostosis syndromes are caused by mutations in genes encoding FGFR-1, FGFR-2, and FGFR-3, and the transcription factors TWIST1 (upstream regulator of FGFRs), MSX2, and ALX4. Gain-of-function mutations are associated with the human genes MSX2 , ALX4 and FGFR , whereas loss of function is associated with the TWIST1 gene EFNB1 . In 2016, SIX2 was found in patients with syndromic complex or sagittal synostosis. ZIC1 is among the latest identified craniosynostosis-associated genes. New mutations are expected to be found in the future, on account of which the number of syndromic craniosynostoses will increase. After the discovery of P250R FGFR3 mutation, Muenke syndrome is the most frequent syndromic craniosynostosis, followed by Crouzon syndrome and Pfeiffer syndrome. Apert syndrome ( FGFR2 ) has the lowest frequency. A complete list of genetic causes of craniosynostosis can be found in the OMIM database.
Syndromic craniosynostoses exhibit variable clinical and genetic heterogeneity. Crouzon and Pfeiffer both have their mutation on FGFR2 . These syndromes can occur clinically identical regarding facial features, but in Pfeiffer also broad and short big toes and thumbs are seen. This distinction can be difficult and even irrelevant.
Diagnostics
Targeted FGFR1 , FGFR2 , FGFR3 , and TWIST1 genetic testing is appropriate as a first-line test for patients with coronal, complex nonsyndromic, and syndromic craniosynostosis. Mutation analyses of TCF12 and ERF are justified in all patients with coronal synostosis who are negative for FGFR and TWIST1 mutations. The value of these tests for patients with sagittal and lambdoid CS is minimal, if any.
Most Common Syndromes
Muenke Syndrome
Genetics
- •
F GFR3 Pro250Arg mutation
- •
Autosomal dominant or de novo
- •
Birth prevalence 1:30,000, 8% of the craniosynostosis cases.
Clinical Features
- •
Brachycephaly due to fused (bi)coronal ± other sutures
- •
Various minor anomalies
- •
Mild midface hypoplasia, downslanting palpebral fissures, mild ptosis, high arched palate, strabismus.
- •
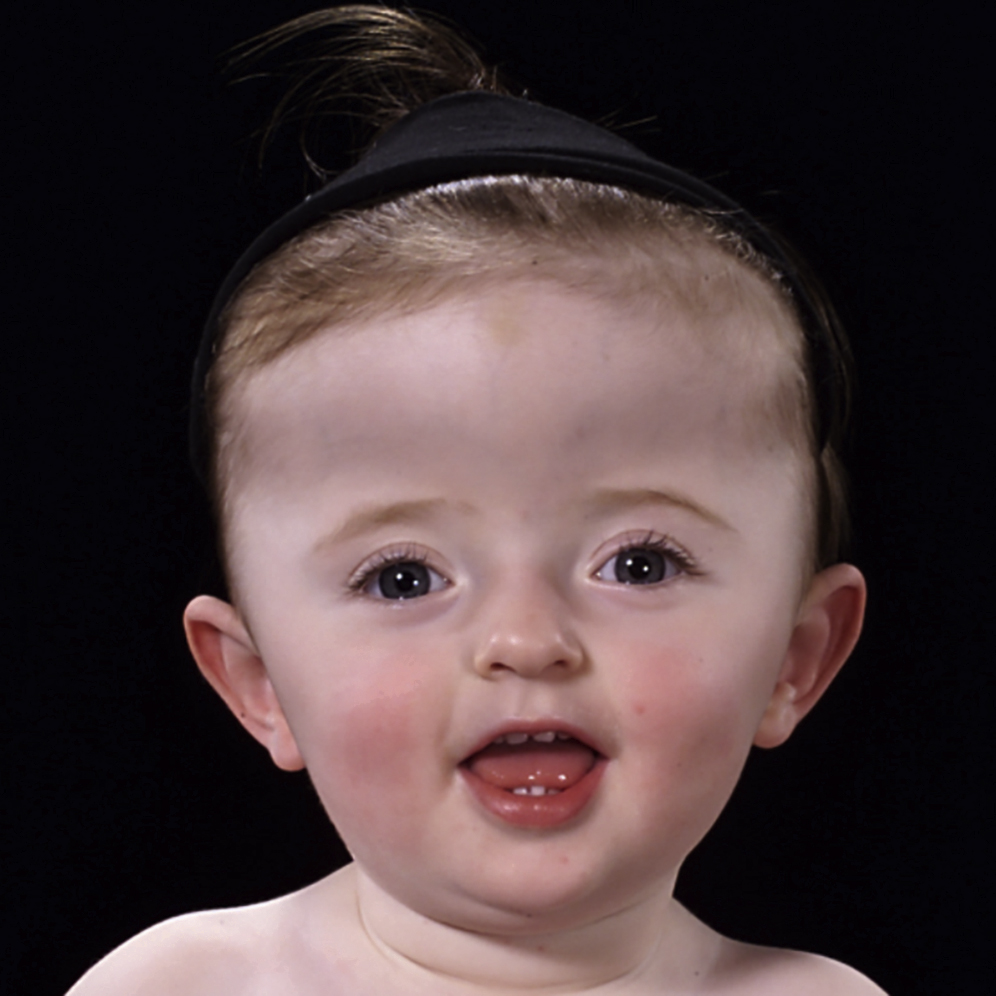
Minor anomalies of hands and feet, e.g., brachydactyly, clinodactyly, can occur. Almost all patients have hearing loss. Typically, females are affected more severely. In general, Muenke patients have a normal intellect, although developmental delay occurs in approximately one-third ( Fig. 20.2 ).
Crouzon Syndrome
Genetics
- •
FGFR2 mutations
- •
FGFR3 mutations Crouzon with acanthosis
- •
Autosomal dominant or de novo (increased risk with older paternal age)
- •
16.5 per million births, 4.8% of the craniosynostosis cases.
Clinical Features
Brachycephaly due to fusion of bicoronal ± sagittal and lambdoid sutures (can be absent or delayed)
- •
Maxillary hypoplasia
- •
Hypertelorism due to short, wide anterior cranial base
- •
Exophthalmus with retruded supraorbital, infraorbital, and lateral orbital rims
- •
Orbital dystopia and strabismus can occur
- •
20% develop optic atrophy
- •
- •
Class III malocclusion.
Up to 30% present with hydrocephalus. Some have a reduced nasopharynx, which can contribute to obstructive sleep apnea (OSA). Also raised intracranial pressure (ICP) and hearing loss can be observed ( Fig. 20.3 ).

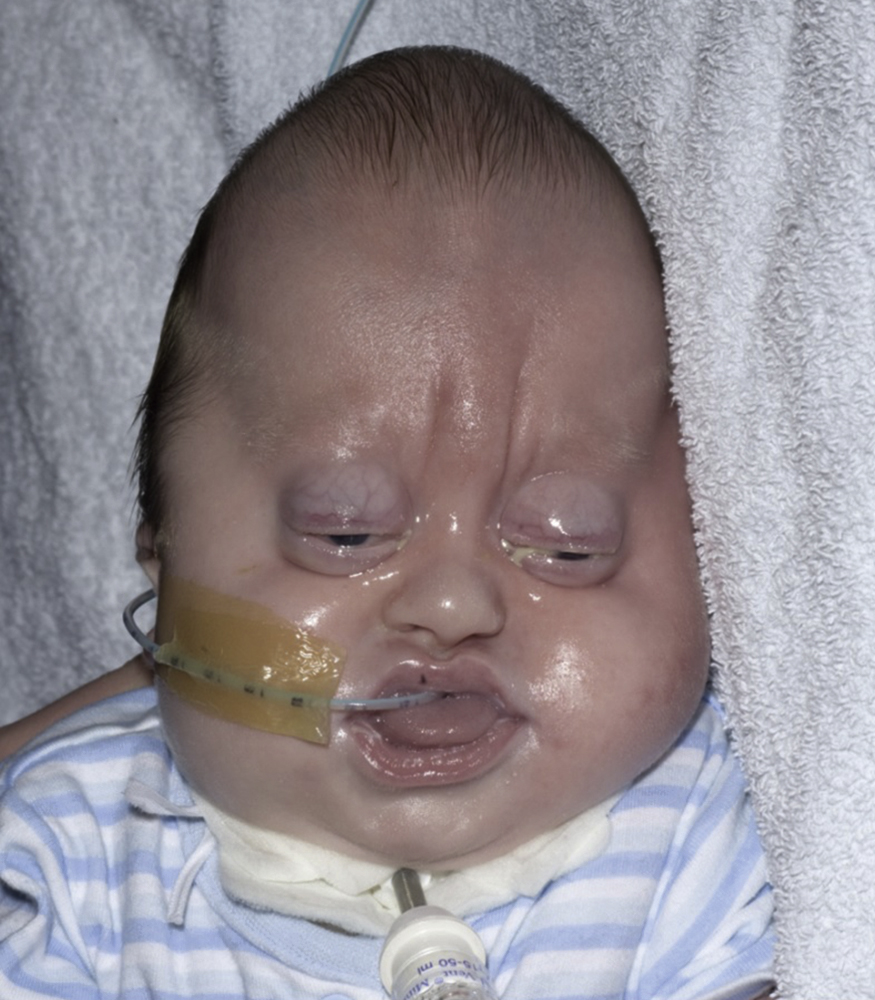
Pfeiffer Syndrome
Genetics
- •
FGFR1 mutation (8p11.2-p11)
- •
FGFR2 mutation (10q26.13)
- •
Autosomal dominant or de novo (increased risk with older paternal age)
- •
1 in 100,000 individuals.
Clinical Features
- •
Brachycephaly due to fused bicoronal sutures
- •
Maxillary hypoplasia
- •
Broad radially deviated thumbs and big toes
- •
Exorbitism due to shallow orbits.
Partial syndactyly of fingers and toes is seen; additional anomalies can include ankylosis of the elbow and other joints, congenital airway malformations, deafness and neurodevelopmental abnormalities ( Fig. 20.4 ).
Apert Syndrome
Genetics
- •
FGFR2 (10q26.13) mutations
- •
Amino acids Ser252Trp and Pro253Arg
- •
Deletion exon IIIc
- •
Alu insertion exon IIIc
- •
- •
Autosomal dominant or de novo (increased risk with older paternal age)
- •
Birth prevalence 15.5 per 1 million births, 4.5% of craniosynostosis cases.
Clinical Features
- •
Turribrachycephaly due to fused bicoronal sutures
- •
Hypertelorism
- •
Exorbitism due to shallow orbits
- •
Downslanting palpebral fissures
- •
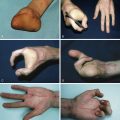
Complex syndactyly of the hands and feet (Upton classification, see Table 20.1 )
TABLE 20.1
Upton’s Classification of the Apert Hand (1991)
Deformity
Type I
Type II “Mitten Hand”
Type III “Hoof Hand”
Thumb radial clinodactyly
Present
Present
Present
Index radial clinodactyly
Absent
Present
Present
First web syndactyly
Simple (nonosseous)
Simple (nonosseous)
Complex (osseous)
Complex 2–3–4 syndactyly and symbrachyphalangism
Present
Present
Present
4–5 syndactyly
Simple, incomplete
Simple, incomplete
Simple, complete
- •
Maxillary hypoplasia
- •
Wide-open calvarial defect from the root of the nose to the posterior fontanelle in the midsagittal plane
- •
Class III malocclusion, anterior open bite and dental crowding
- •
Cleft palate with high arched palate can occur.
Additional anomalies can include fusion and malformation of other joints, e.g., the elbows and shoulders, cervical vertebral fusions, congenital airway malformations, deafness, and neurodevelopmental abnormalities. Also, globe subluxation can occur. The quality of the skin frequently varies from normal, to dermal problems such as acne and hyperhidrosis ( Fig. 20.5 ).

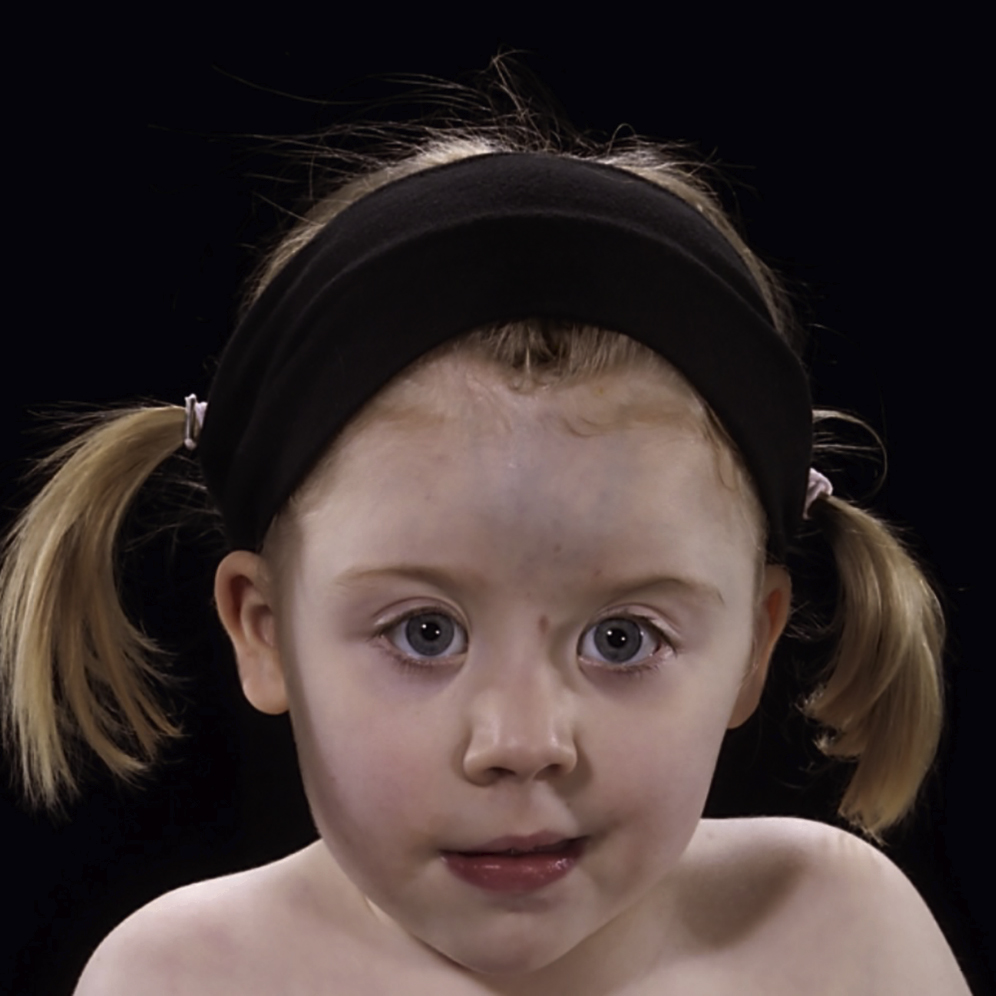
Saethre–Chotzen Syndrome
Genetics
- •
TWIST1
- •
FGFR2
- •
Autosomal dominant or de novo.
Clinical Features (see Fig. 20.6 )
- •
Brachycephaly due to fused (usually) uni- or bicoronal (sometimes metopic) sutures
- •
Facial asymmetry
- •
Hypertelorism
- •
Maxillary hypoplasia
- •
High forehead and low frontal hairline, and late closing fontanelle
- •
Strabismus, ptosis, lacrimal duct stenosis, deviated nasal septum
- •
Small low-set posteriorly rotated ears with prominent crus
- •
Limb anomalies including: radioulnar synostosis, brachydactyly, cutaneous syndactyly and hallux valgus
- •
Congenital heart defects
- •
Ptosis
- •
Small ears with prominent horizontal crurae.
Rare Syndromes
Craniosynostosis occurs in over 200 known syndromes and the majority occur extremely rarely. A few examples are: craniofrontonasal syndrome, ERF , TCF12 , Antley–Bixler syndrome with or without genital anomalies and disordered steroidogenesis, Baller–Gerorld syndrome, Beare–Stevenson syndrome, Carpenter syndrome, Jackson–Weiss syndrome, Loeys–Dietz syndrome, and Shprintzen–Goldberg syndrome.
Parameters of Care for Craniosynostosis
Over the years, a multidisciplinary approach for children with craniosynostosis has evolved. Various specialties are involved for optimal diagnosis, management, and treatment. Interdisciplinary craniofacial teams are commonly composed of members with expertise in anesthesia, craniofacial surgery, dentistry, genetics, hand surgery, intensive care, neurosurgery, nursing, ophthalmology, oral and maxillofacial surgery, orthodontics, otolaryngology, pediatrics, prosthodontics, psychology, public health, radiology, social work and speech-language pathology. Care for patients with craniofacial anomalies is best provided in a Craniofacial Team because of the complexity of the cases. Patients with craniosynostosis can have and develop significant problems and might need timely management. Therefore, it is important to assess a child with craniosynostosis as early as possible.
Craniofacial Assessment
Prenatal Diagnosis
Most patients with craniosynostosis are not detected during pregnancy. From the start of formation of cranial sutures at about 16 weeks’ gestation and the antenatal ultrasound at 20 weeks’ gestation there has not been enough time for growth distortion of the skull to have occurred, except in the most severe cases.
Initial History and Examination from Birth to Age 3 Months
In the case of a patient suspected for craniosynostosis, craniofacial assessment should be performed. The most common presentation is an unusual head shape. The head may be long and narrow (scaphocephaly, dolichocephaly), triangular at the front (trigonocephaly), broad and flattened (brachycephaly) or skewed (plagiocephaly). The aims of a craniofacial assessment are to determine: (1) whether craniosynostosis is present; (2) whether there are additional features suggesting an associated syndrome; and (3) to assess whether urgent or elective management is required. The management of craniosynostosis varies because the cause and presentation of craniosynostosis is very heterogeneous. Most isolated nonsyndromic cases present electively, but a minority of syndromic cases present acutely and require immediate intervention.
During this assessment, information should be gathered on possible identifiable causes: (1) intrauterine constraint, e.g., primiparity, multiple pregnancy, abnormal lie, oligohydramnios; (2) teratogenic exposure, e.g . , maternal use of oral retinoids, sodium valproate or phenytoin during pregnancy; (3) family history of abnormal head shape and genetic abnormalities. Moreover, information should be collected on: the conception (normal or assisted), pregnancy, birth history, and birth weight of the patient and their growth. Possible functional issues should be addressed and questions must be asked on the airway (breathing difficulties), feeding (choking, vomiting on feeds), eye protection (failure of eyelids to cover eyes during sleep or irritability), and raised ICP, because these problems might be an indication for early and acute intervention.
On examination, the skull should be examined from the front, back, sides, and top as well as palpation of the sutures for ridging (associated with suture fusion), assessment of the size, shape, and tension of the fontanelles, and head circumference measurements. The cephalic index (ratio of maximum breadth to length of skull, normally between 0.76 and 0.83 (in boys) or 0.84 (in girls)) provides an objective measurement. Moreover, signs for other congenital anomalies should be looked for in areas, such as the face, including mouth, e.g., hyper- or hypotelorism, exorbitism, midface hypoplasia, facial asymmetry, ear size, ear position and shape, cleft palate and malocclusion; hand and feet, e.g., syndactyly, longitudinally split nails, deviated thumb or big toe.
Other examinations should include:
- •
Eye evaluation (funduscopic examination and visual evoked potentials)
- •
ENT and hearing assessment
- •
Genetics assessment
- •
General assessment by a pediatrician (e.g . , cardiovascular system)
- •
Speech and language evaluation
- •
Photography: clinical 2D (and if possible 3D) photographs
- •
Radiological image: X-ray/CT/MRI as necessary
- •
Assessment of parents for suggestive features of craniosynostosis.
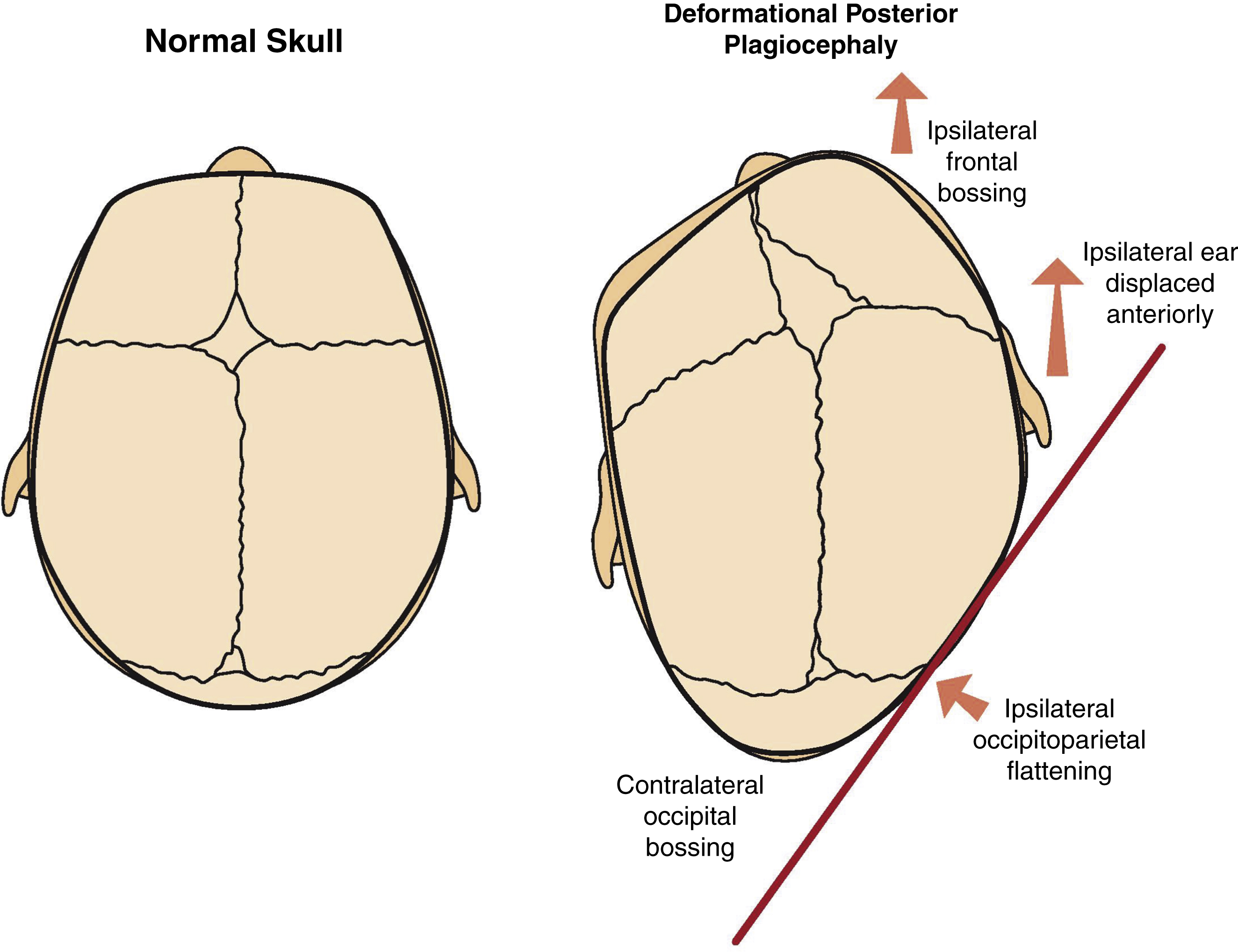
Deformational Posterior Plagiocephaly
Craniosynostosis must be differentiated from deformational plagiocephaly that can occur due to prenatal, perinatal, and/or postnatal external causes.
Prenatal
Deformational plagiocephaly can occur due to prematurity, or intrauterine constraint, such as twin pregnancy.
Perinatal
Deformational plagiocephaly can occur due to assisted delivery with, for example, the use of a forceps.
Postnatal
Deformational plagiocephaly has become increasingly common after the “Back to Sleep” campaign. In 1992, the American Academy of Pediatrics (AAP) Task Force on Infant Positioning and Sudden Infant Death Syndrome (SIDS) recommended a back or side position of the infant when laid to sleep to reduce the incidence of SIDS. The incidence decreased from 1.2/1000 live births in 1992 to 0.56/1000 in 2001. , However, this resulted in a deformation of the normal skull. The skull of a newborn is malleable and due to pressure on the side the infant lies, changes in head shape can occur, including occipitoparietal flattening on the side where the child tends to lie, ipsilateral frontal bossing and ipsilateral anterior displacement of the ear and contralateral occipital bossing ( Fig. 20.7 ).