36 Craniofacial microsomia
Synopsis








Basic science/disease process
Etiopathogenesis
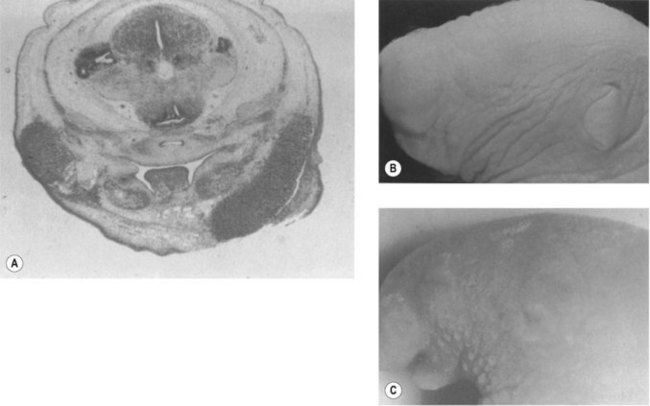
The underlying cause or etiopathogenesis of CFM remains a subject of debate. The prevailing theory was that CFM was a sporadic event, possibly precipitated by exposure to teratogens. Other studies have instead suggested a fundamental role for genetic transmission in some patients. The etiology of CFM is probably heterogeneous among individuals, with variable contributions from extrinsic and intrinsic factors. Support for a teratogenic etiology of CFM is based largely on animal studies. Poswillo exposed mouse embryos to triazine by maternal administration of the drug, causing CFM-like phenotypes (Fig. 36.1). Focal hematomas arising from disruption of the stapedial artery were also observed. Although a “stapedial artery hemorrhage etiology” is attractive because the vessel is a second branchial arch derivative, a causative association between the bleeding and the deformities has not been made. The hemorrhages occurred 14 days after administration of the teratogen, and there was no clear temporal relationship between the hemorrhage appearance and the associated phenotypic deformity. When mice are exposed to triazine later in development (10 days of gestation), all animals developed deformities; however, only a third showed evidence of a hematoma. The authors concluded that triazine has a direct teratogenic effect and the stapedial artery findings were simply a side-effect. In contrast to those described by Poswillo, these animals demonstrated more evidence of bilateral deformities and inner-ear anomalies. Intermittent occlusion of the internal carotid system of fetal sheep late in gestation has been shown to result in deformities similar in appearance to CFM. The vascular disruption hypothesis, therefore, cannot be excluded.
Embryology
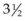 -week-old human embryo as a thickening of the ectoderm in the side of the head – the otic placode. This area is enfolded to become the otic pit and is subsequently pinched off to become the otocyst. By means of a series of folds, the otocyst differentiates in the 3-month-old fetus into the endolymphatic duct and sac, the semicircular endolymphatic ducts, the utricle, the saccule, and the organ of Corti. By the fifth month of fetal life, the sensory end organ of the ear attains adult form and size as the cartilaginous otic capsule ossifies.
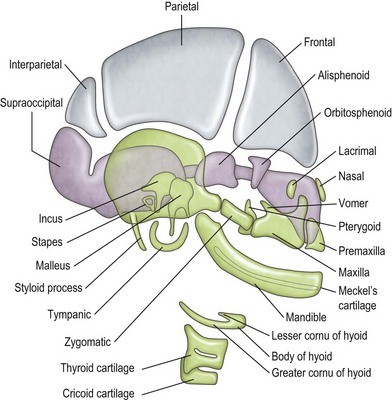
-week-old human embryo as a thickening of the ectoderm in the side of the head – the otic placode. This area is enfolded to become the otic pit and is subsequently pinched off to become the otocyst. By means of a series of folds, the otocyst differentiates in the 3-month-old fetus into the endolymphatic duct and sac, the semicircular endolymphatic ducts, the utricle, the saccule, and the organ of Corti. By the fifth month of fetal life, the sensory end organ of the ear attains adult form and size as the cartilaginous otic capsule ossifies.The mandible, incus, and malleus develop from the cartilage of the first branchial arch (Meckel cartilage) (Fig. 36.2). The stapes (with the exception of the footplate, which originates from the otic capsule), styloid process, and hyoid bone develop from the cartilage of the second branchial arch (Reichert cartilage). The large area of the tympanic membrane, connected by the lever system of the ossicular chain to the small area of the oval window, provides the ear with an effective mechanism to overcome the sound barrier between air and water.
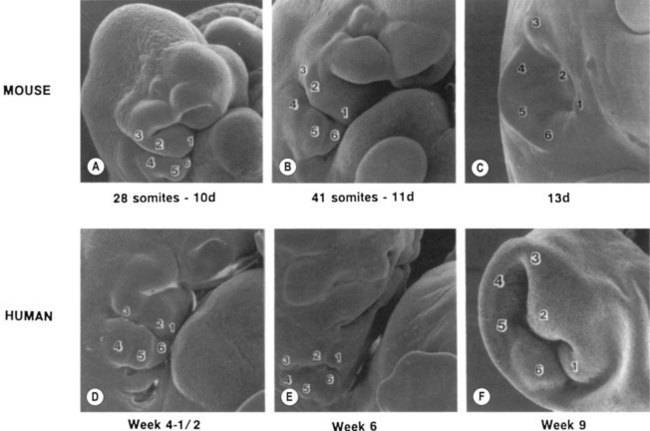
By the third fetal month, the external auricle has been formed from the first and second branchial arches on either side of the first branchial groove, which is the primary shallow, funnel-shaped external auditory meatus (Fig. 36.3). From the inner end of the primary meatus, a solid cord of ectodermal cells extends farther inward, with a bulb-like enlargement adjacent to the middle ear. It is not until the seventh fetal month that the cord canalizes, beginning medially to form the tympanic membrane and extending laterally to join with the primary meatus to form the completed external auditory meatus. The external and middle ears, although capable of transmitting sound to the inner ear, are not yet of adult form and size.
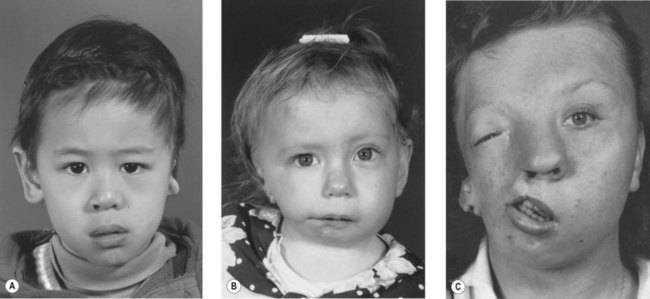
Pathology
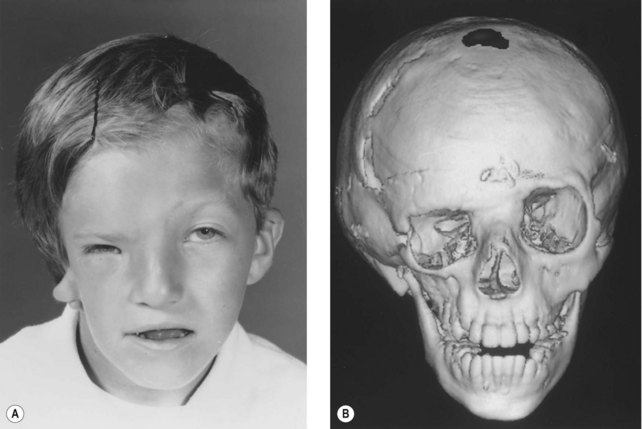
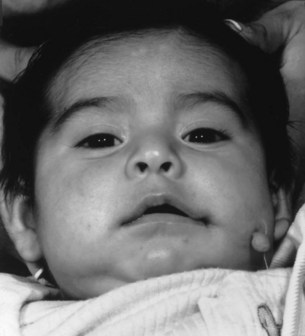
Whereas the jaw and ear deformities are the most conspicuous in the majority of patients, the first and second branchial arches and the structures derived from them are intimately interlinked with the chondrocranium and membranous bones of the skull; associated deformities of the temporal bone and other cranial bones are inevitable. In extreme forms of the dysplasia, widespread craniofacial involvement is evident (Fig. 36.4). As Pruzansky stated, maldevelopment in one area may trigger a “domino effect,” with involvement of the entire craniofacial skeleton including microphthalmos, orbital dystopia, and orbitofacial clefts.
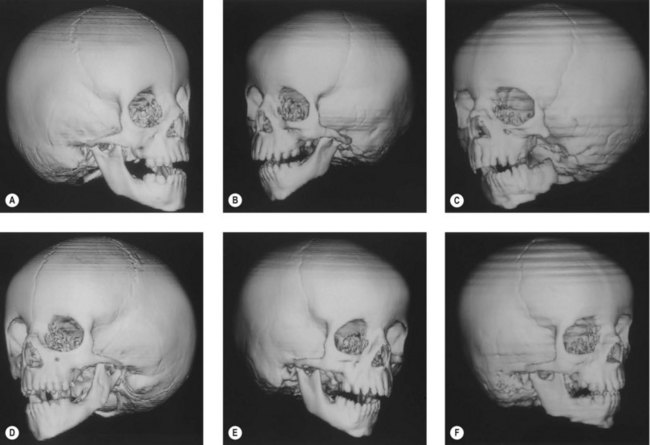
Skeletal tissue
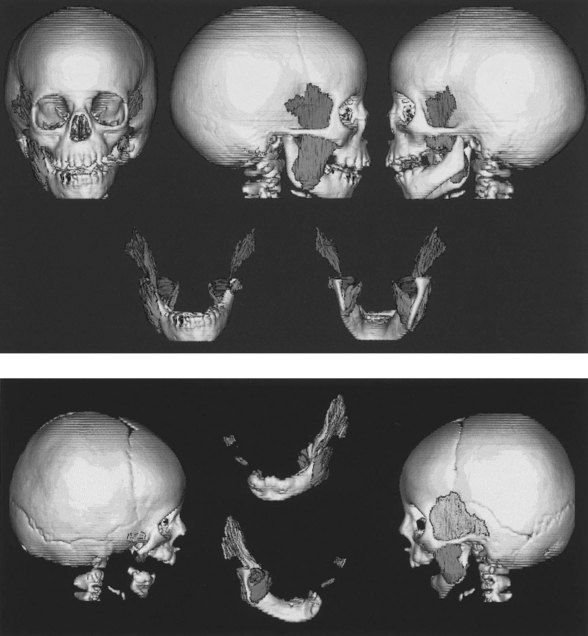
Ramus and condyle malformations vary from minimal hypoplasia or blunting of the condyle to its complete absence in association with hypoplasia or agenesis of the ramus (Fig. 36.5). In all patients, condylar anomalies can be demonstrated, and this finding may be pathognomonic of the syndrome. As a consequence, the spatial relationships of the malformed or deficient skeletal parts, as well as the associated neuromuscular components, become of paramount importance in the diagnosis and planning of treatment.
Mandibular growth deficiency usually is closely related to the degree of hypoplasia of the condyle. In more severe conditions there is considerable disparity in condylar growth between the affected and contralateral sides. The cant of the occlusal plane (higher on the affected side) is caused by the short, hypoplastic ramus and by hypoplasia of the ipsilateral maxillary dentoalveolar process (Fig. 36.6). The floor of the maxillary sinus and of the nose on the affected side is canted at a higher level. In some patients, the base of the skull is elevated on an inclined plane similar to the inclined occlusal plane. Anteroposterior and superoinferior dentoalveolar and skeletal dimensions are reduced on the affected side. Crowded dentition, with a characteristic tilt of the anterior maxillary and mandibular occlusal planes upward on the affected side, is often noted.

Craniofacial bones other than the mandible or maxilla can be involved, especially the tympanic and mastoid portions of the temporal bone; the petrous portion usually is remarkably spared. The styloid process is frequently smaller on the affected side. The mastoid process can have a flattened appearance, and there can be partial or complete lack of pneumatization of the mastoid air cells (Fig. 36.7).
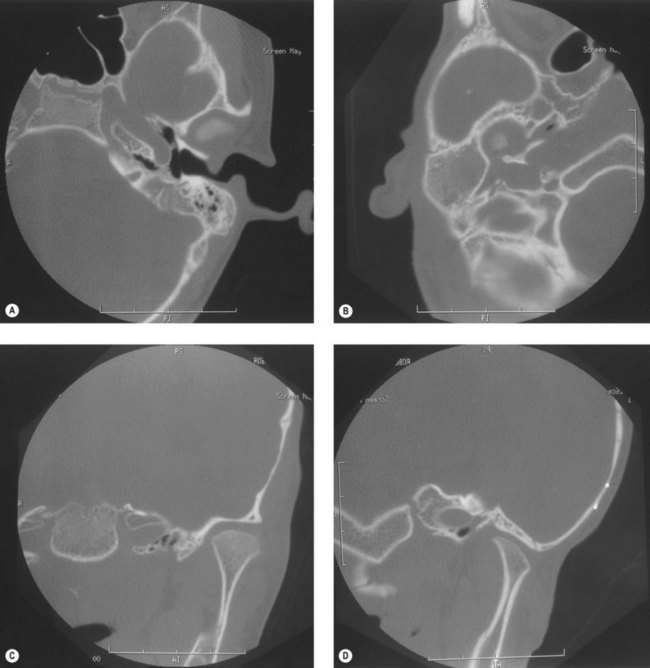
Disparities in the vertical axis of the orbit can be seen, with or without evidence of microphthalmos (Fig. 36.8). Often in this situation, there is flattening of the ipsilateral frontal bone – an appearance of plagiocephaly without radiographic evidence of coronal synostosis.
Nonskeletal (soft) tissue
The “functional matrix” theory of Moss attributes the overall growth and development of the head to the development of the soft-tissue matrix and functional spaces. The matrix is composed of cells, tissues, organs, and air volumes that serve a functional role. The associated “hard” tissues, such as bone and cartilage, serve to protect and support the functional matrix. Their morphology is solely determined by the functional matrix. As summarized by Moss, “bones do not grow, they are grown.” This theory has been supported by animal research examining the effect of transposition of muscles of mastication on bone morphology. Investigation of the effect of soft tissue on bone shape in humans with unilateral CFM has been limited to computed tomographic (CT) analysis. Results of these studies do suggest that changes in the muscles of mastication can elicit a postnatal change in bone morphology but that the opposite – bone changes affecting muscle – does not take place (Fig. 36.9). It can be speculated that, if the functional matrix theory is validated, future treatment of CFM may be limited to early manipulation of the soft-tissue matrix to elicit a secondary effect on the associated bone.
Ear
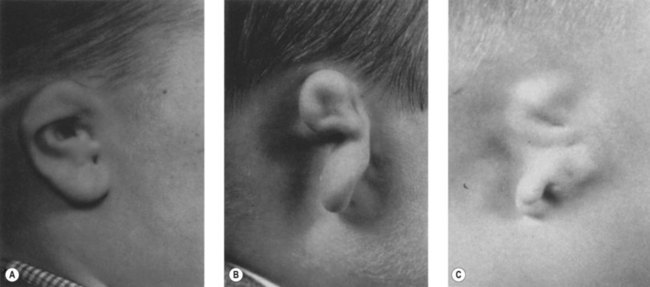
Auricular malformations are a usual manifestation of the syndrome. Meurman proposed a classification of the auricular anomalies based on the studies of Marx (1926): grade I, distinctly smaller malformed auricles with most of the characteristic components; grade II, vertical remnant of cartilage and skin with a small anterior hook and complete atresia of the canal; and grade III, an almost entirely absent auricle except for a small remnant, such as a deformed lobule (Fig. 36.10).
Skin and subcutaneous tissue
In the series of patients described by Grabb, 10% had malformations of the eyes and eyelids or palate. Transverse facial clefting, ranging from macrostomia to a full-thickness defect of the cheek, can be present (Fig. 36.11). The clefts probably result from a failure of the maxillary and mandibular processes to fuse. In embryonic development, the lateral commissure of the oral fissure is initially situated at the point of bifurcation of the maxillary and mandibular processes. With fusion of these and development of the muscles of mastication, the original broad mouth is reduced in size. In addition, the parotid glands, originally located near the embryonic oral commissure, grow laterally toward the developing ear, but the parotid duct papillae remain in their more medial position.
Extracraniofacial anatomy
Horgan et al. reviewed 121 cases of CFM and reported that 67 patients (55%) had at least one extracraniofacial (vertebral, cardiac, genitourinary) anomaly, with some having up to seven malformations. They also identified a relationship between the presence of these malformations and the severity of the craniofacial bone and soft-tissue involvement. The spectrum and incidence of associated malformations are listed in Table 36.1.