33 Craniofacial clefts
Synopsis








Introduction
Congenital craniofacial clefts are abnormal disfigurements of the face and cranium with deficiencies, excesses, or even a normal (but separated) amount of tissue occurring along linear regions.1–4 Of all congenital facial anomalies, craniofacial clefts are among the most disfiguring. They may be seen in a variety of patterns and varying degrees of severity. Although at first glance they appear to defy definable patterns, most craniofacial clefts occur along predictable embryologic lines.5 Craniofacial clefts may be either unilateral or bilateral and one cleft type can manifest on one side of the face, while a different type is present on the other side.
Classifications of craniofacial clefts
Craniofacial malformations are rare, and have multiple variations and a spectrum of severity. For diagnostic and treatment purposes, the use of similar terminology describing embryologic maldevelopment, genetic etiology, or anatomic landmarks is beneficial. Organization of the seemingly heterogeneous clefting malformations is necessary for both morphogenetic understanding and knowledge of surgical anatomy for treatment. Therefore, orderly systems of classification have been described.1–4
The American Association of Cleft Palate Rehabilitaion (AACPR) separated craniofacial clefts into four categories based on pathologic location: (1) mandibular process clefts; (2) naso-ocular clefts; (3) oro-ocular clefts; and (4) oroaural clefts.6 First, mandibular process clefts group malformations of the mandible and lower lip. Second, naso-ocular clefts include malformations located between the nasal ala and the medial canthus. Third, oro-ocular clefts consist of clefting anomalies connecting the oral cavity to the orbit between the medial and lateral canthus. Fourth, oroaural clefts describe anomalies involving the region from the oral commissure to the auricular tragus. Subsequent to the AACPR classification, Boo-Chai made modifications based on surface anatomic landmarks, including skeletal components.7 The oro-ocular clefts were subdivided into two types, the oromedial canthus and orolateral canthus, with the infraorbital foramen as a reference point.
The Karfik classification has an embryologic and morphologic basis and is divided into five groups: (1) group A: rhinencephalic malformations; (2) group B: anomalies of the first and second branchial arch; (3) group C: orbitopalpebral malformations; (4) group D: craniocephalic malformations (e.g., Apert and Crouzon syndrome); and (5) group E: atypical deformities from congenital tumors, atrophy, hypertrophy, and true oblique clefts which cannot be related to any embryologic fusion line.8 Group A, the rhinencephalic malformations, was further divided into two subtypes: group A1, axial malformations derived from frontonasal promenince, and group A2, paraxial malformations adjacent to the nasal region. Group B, anomalies of the first and second branchial arch, was also further divided into two subtypes: group B1, composed of the lateral otocephalic malformations (craniofacial microsomia, Treacher–Collins syndrome, Pierre Robin sequence, and auricular malformations) and group B2, including the mandibular midline malformations.
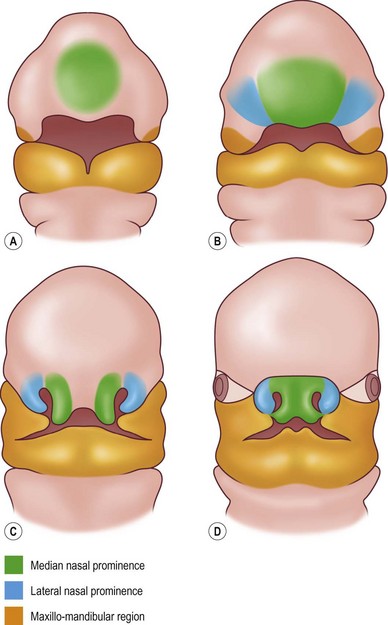
The Van der Meulen classification used the term “dysplasia” since some of the malformations did not represent true clefts.9 The defects were labeled by the name of the developmental area (facial processes and bones) that is involved. The malformations are believed to occur before or during the fusion of the facial processes, but before the start of ossification (Fig. 33.1).
Median craniofacial clefts require special consideration because of variation in malformations and deformations. With tissue agenesis and holoprosencephaly at one end (the hypoplasias), and frontonasal hyperplasia and excessive tissue (the hyperplasias) at the other end, median anomalies with normal tissue volume occupy the middle portion of the spectrum.10 Noordhoff et al. limited the term “holoprosencephaly” to cases of alobar brain and preferred the term “dysplasia”rather than “dysgenesis.”11 Thus, for clarification, median craniofacial dysplasias have been organized and divided into:
I: median craniofacial hypoplasia (tissue deficiency or agenesis)
II: median craniofacial dysraphia (normal tissue volume but clefted)
III: median craniofacial hyperplasia (tissue excess or duplication).
Under each division further subclassification is used to describe the specific anomalies of the number 0–14 clefts (Table 33.1).
Table 33.1 Classification of median craniofacial dysplasia
| Subclassification | Description |
|---|---|
| I. Median craniofacial hypoplasia | Tissue deficiency |
| A. Holoproencephalic spectrum (alobar brain) | Single holistic brain with midline facial hypoplasia or agenesis (Fig. 33.1). Four subclassifications: cyclopia, ethmocephaly, cebocephaly, primary palate agenesis |
| 1. Cyclopia | 1. Single eye in a single orbit, arrhinia with proboscis often located above the single orbit and microcephaly (Fig. 33.2) |
| 2. Ethmocephaly | 2. Severe hypotelorism but separate orbits. Arrhinia with proboscis located between the orbits (Fig. 33.3) |
| 3. Cebocephaly | 3. Severe hypotelorism. Proboscis-like rudimentary nose (Fig. 33.4) |
| 4. Primary palate agenesis | 4. Premaxillary segment missing or hypoplastic (Fig. 33.5) |
| B. Median cerebrofacial hypoplasia (lobar brain) | Separate lobes to brain but with midline cerebral malformation; midline facial hypoplasia (Fig. 33.6) |
| C. Median facial hypoplasia | Midline facial hypoplasia without gross cerebral involvement (Fig. 33.7) |
| D. Microforms of median facial hypoplasia | 1. Binder anomaly (maxillonasal dysplasia) (Fig. 33.8) |
| 2. Central maxillary incisor anomaly (Fig. 33.9) | |
| 3. Absent upper lip frenulum (Fig. 33.10) | |
| II. Median craniofacial dysraphia | Normal tissue volume but clefted |
| A. True median cleft | Isolated cleft of the upper lip or abnormal split between the median globular process. It can be an incomplete (Fig. 33.11) or complete form (Fig. 33.12) |
| B. Anterior encephalocele | Cystic malformation in which central nervous structures are abnormally displaced or herniated through a defect in the cranium (Figs 33.3 and 33.14) |
| III. Median craniofacial hyperplasia | Tissue excess or duplication. All forms of excess tissue starting from just thickened or duplicated nasal septum to the more severe forms of frontonasal dysplasia (Figs 33.15 and 33.16) |
I: Median craniofacial hypoplasia (tissue deficiency and/or agenesis)
A Holoprosencephalic spectrum (alobar brain)
1. Cyclopia: described as a single eye in a single orbit. Arrhinia exists with proboscis often located above the single orbit. Microcephaly is also a component.
2. Ethmocephaly: severe hypotelorism exists but the orbits are separate anatomically. Again, arrhinia is present; however, the proboscis is located in between the orbits.
3. Cebocephaly: moderate to severe hypotelorism is noted. There is a proboscis-like rudimentary nose in a more typical nasal position.
4. Primary palate agenesis: the primary palate, including the premaxillary segment and associated midline structures, is absent or severely deficient. Hypotelorism is seen.
D Microforms of median facial hypoplasia
1. Binder syndrome or anomaly (maxillonasal dysplasia) patients have characteristic flat nasomaxillary facial region with deficient or absent nasal spine and a negative overjet from class III anterior incisor relationship.
2. Abnormalities of the maxillary central incisors: three variants of this include:
II: Median craniofacial dysraphia
B Anterior encephaloceles
Anterior encephaloceles are divided into frontoethmoidal and basal groups. In frontoethmoidal encephaloceles, the defect occurs at the junction of the frontal and ethmoidal bones (the foramen cecum).12 Nasoethmoidal encephalocele is considered a Tessier number 14 cleft or frontonasal dysraphia in Mazzola’s morphological classification. Basal encephaloceles are associated with a defect at or behind the crista galli and, in some cases, they may protrude through a defect in the sphenoid bone and are called transsphenoidal encephalocele.13
III: Median craniofacial hyperplasia (tissue excess or duplication)
“Frontonasal dysplasia” was the most widely known of these types of hyperplasias.14 Objections are raised about the terminology of this condition. Typically, the term “dysplasia” refers to the whole spectrum of abnormal tissue development starting from tissue agenesis and hypoplasia all the way to the other extreme of hyperplasia and excess tissue.
The basic defect of median craniofacial hyperplasia is not known. Embryologically, if the nasal capsule fails to develop properly, the primitive brain vesicle fills the space normally occupied by the capsule, thus producing anterior cranium bifidum occultum and leading to morphokinetic arrest in the positioning of the eyes and nostrils, which tend to maintain their relative fetal positions.15,16 Experiments have shown that a reduction in the number of migrating neural crest cells results in these multiple defects.17,18
Other nasal findings may range from a notched broad nasal tip to completely divided nostrils with hypoplasia and even absence of the prolabium and maxilla with a median cleft lip. In addition, variable notching of the ala is described. Occasionally associated abnormalities include accessory nasal tags, low-set ears, conductive hearing loss, mild to severe retardation, basal encephalocele, and agenesis of the corpus callosum. Importantly, a high incidence of ocular abnormalities is described. More distant anomalies include tetralogy of Fallot, absence of the tibia, and others. When hypertelorism is severe or when extracephalic anomalies occur, mental deficiency appears to be more likely and more severe.14,15,17
The Tessier classification of craniofacial clefts was described in 1976.1 This classification has proven to be the most complete and has withstood the test of time. This insightful classification was based on the extensive personal experience by Tessier in the anatomy laboratory, in the operating room, and from his embryologic knowledge. The terminology is uniform and the characterizations of specific features are detailed. It is also reproducible by clinicians evaluating craniofacial clefts. In addition, the classification links the clinical observations with underlying skeletal deformity documented by preoperative three-dimensional (3D) computed tomography (CT) scan imaging and confirmed during surgery. Correlation of the clinical appearance with the surgical anatomic findings improves the clinical utility of this system for the craniofacial surgeon. Distinctive features such as Tessier craniofacial clefts are described below.
Epidemiology and etiologic factors
The true incidence of craniofacial clefts is unknown because of their rarity and because of the difficulty in recognizing sometimes subtle physical findings in mild malformations. However, the incidence of craniofacial clefts has been estimated at 1.4–4.9 per 100 000 live births.1–3 The incidence of rare craniofacial clefts compared to common cleft lip and palate malformations may range from 9.5 to 34 per 1000.3
Most rare craniofacial clefts occur sporadically. However, the role of heredity in the causation of rare craniofacial clefts seems to occur in Treacher–Collins syndrome and in some familial cases of Goldenhar syndrome. A dominant gene defect (TCOF-1) causes Treacher–Collins syndrome.19 Although penetrance is somewhat variable, the malformation is very consistent. In a TCOF-1 gene knock-out animal model, regional massive cell death affected crest cell mesenchymal migration and resulted in zygomatic abnormalities. Constriction limb deformities (amniotic band syndrome) have also been associated with rare facial clefting. Coady et al. found a statistically significant association between craniofacial clefts and limb ring constrictions.20
Based on animal and human clinical studies, many environmental factors have also been shown to cause facial clefts. These investigations produced four major categories: (1) radiation21,22; (2) infection23,24; (3) maternal metabolic imbalances25; and (4) drugs and chemicals.26 A considerable number of drugs and chemicals have teratogenic potential but few have been shown to cause craniofacial malformations in humans. Some medications, including those containing retinoic acid, are being looked at as a cause of facial malformations.27
Embryologic craniofacial development
The three primary germ layers, ectoderm, mesoderm, and endoderm, are the basis for tissue and organ formation.28 In the third week of gestation, the primitive tissues of the trilaminar embryo give rise to notochordal and prechordal mesoderm. Simultaneously, the rostral ectoderm differentiates to form highly specialized neural crest cells, which are responsible for the ultimate development of the brain and midline facial structures.29 The ectoderm forms a neural plate with bilateral folds which conjoin into a neural tube. During closure of the neural tube, neural crest cells (mesenchyme) migrate into underlying tissue, forming pluripotential stem cells. The embryonic prominences of the face are formed by the migration of these neural crest cells. A segmental pattern of ventral migration of neural crest, termed rhombomeres, provides the precursors of cartilage, bone, muscle, and connective tissue of the face and head.
Any defect in the quantity and quality of this migrating ectomesenchyme is manifest as a craniofacial malformation from severe holoprosencephaly to minor clinical stigmata of craniofacial clefts like dimples or skin tags.30 Another cause for arrested development causing dysplasias and dystopias (the abnormal formation and location of structures) is abnormal development or involution of embryologic arteries.31
Starting at the fourth week of gestation, the face assumes a recognizable form.32 Between 4 and 8 weeks of gestation the crown–rump length increases from approximately 3.5 mm to 28 mm. The double-layer stomodeal membrane creates the opening for the primitive mouth. An overhanging frontonasal prominence represents the superior border of the stomodeum.2 Five prominences (the frontonasal and paired maxillary and mandibular) formed by neural crest migration surround the stomodeum (Fig. 33.1). The frontonasal prominence is formed by neural crest cells migrating ventrally from the mesencephalic region and contributes to the frontal and nasal bones. The maxillary and mandibular prominences are formed by more caudally located migrating neural crest cells that encounter pharyngeal endoderm in their ventral migration around the aortic arches.
Optic vesicles appear from lateral envaginations of the diencephalons and induce lens placodes in ectoderm and neural crest migration to form the sclera. Defective optic vesicle formation results in microphthalmos, or anophthalmos. The movement of this optic tissue from lateral to medial results from the narrowing frontonasal prominence and expansion of the lateral face. Inadequate transition of the eyes produces hypertelorism and overmigration produces hypotelorism or even median cyclopia.33
The posterior aspect of each nasal pit is separated from the oral cavity by an oronasal membrane. Failure of this membrane to disintegrate normally leads to choanal atresia.34 The paired median nasal processes merge with the frontonasal prominence to form the majority of the frontal process. These structures gradually enlarge and superiorly displace the frontonasal prominence. During the sixth week, the two median nasal processes coalesce in the midline and their most caudal limbs, the premaxillary prominence, expand above the stomodeum. The nasal tip, philtrum, columella, cartilaginous septum, and primary palate are derived from these paired median elements. Cephalad to the medial nasal process, the frontonasal process persists to form the nasal dorsum and root. Elevation of the lateral nasal prominence creates the nasal alae. Defects during this development may be midline and produce arrhinia or a bifid nose.
The maxillary processes are paired mesodermal masses that lie cephalad to the mandibular arch and ventral to the optic neuroectoderm. These triangular masses enlarge, separate from the mandibular arch, and then migrate ventrally. The maxillary process ultimately coalesces with the mesoderm of the globular processes to form the upper lip. The cheek, maxilla, zygoma, and secondary palate are also derived from the maxillary processes. Between the maxillary prominence and lateral nasal prominence a depression exists with a solid rod of epithelial cells.35 The ends of this rod form a connection from the nasal pit to the conjunctival sac (nasolacrimal duct). With inadequate neural crest cell migration a fissure within the line of this duct may persist as an oblique facial cleft.
Failure of fusion
Two theories exist that describe how embryologic failure or errors result in craniofacial cleft malformations. First, the “fusion failure” theory suggests that clefts are created when fusion of facial processes fail.36 Second, the “failure of mesodermal penetration” theory implicates the lack of mesoderm and neuroectoderm migration and penetration into the bilaminar ectodermal sheets as the cause of craniofacial clefts.37 Although most of the current knowledge is based on animal cleft lip and palate studies, rare craniofacial clefts may be produced by similar mechanisms.
The “failure of fusion” theory, proposed by Dursy in 1869 and His in 1892, purported that the free edges of the facial processes unite in the central region of the face.35 As various processes fuse, the face gradually forms. When epithelial contact is established between opposing facial processes, mesodermal penetration completes the fusion. Dursy suggested that the upper lip is created when finger-like advancing ends of the maxillary process and the paired global process unite. He asserted that disruption of this sequence results in craniofacial cleft anomalies.
Proponents of the mesodermal penetration theory believed that the finger-like ends of the facial processes do not exist. Warbrick38 and Stark et al.and Ehrmann39 suggest that the central facial processes are composed of bilamellar sheets of ectoderm. This bilamellar membrane is bordered by epithelial seams, which delineate the principal processes. During development, the mesenchymal tissue migrates and penetrates this double-layered ectoderm, called the “epithelial wall.” Caudal to the stomodeum, the lower face is formed by the branchial arches. The arches consist of a thin sheet of mesoderm, which lie between the ectodermal and endodermal layers. The neural crest cells of neuroectodermal origin, which arise from the dorsolateral surface of the neural tube, migrate under the ectoderm and supplement the mesoderm of the frontonasal process and branchial arches.40 Most of the craniofacial skeleton is believed to be formed by these neural crest cells. If neuroectoderm migration and penetration do not occur, the epithelium breaks down to form a facial cleft. The severity of the cleft is proportional to the failure of penetration by the neuroectoderm. Unfortunately, the precise nature of the proposed mechanisms in the formation of rare craniofacial clefts is not known. Nevertheless, the concepts of fusion and mesodermal penetration provide an understanding of the problems of the rare craniofacial cleft.
Neuromeric theory
Newer understanding of neuroembryology suggests that a direct relationship exists between the development of the nervous system and those structures to which its contents are dedicated. The neural tube is conceived as a series of developmental zones within the central nervous system.41,42 Six prosomeres provide a cartesian system to organize the tracts and nuclei of the prosencephalon (forebrain). The mesencephalon (midbrain) and rhomboencephalon (hindbrain) are subdivided into two mesomeres and 12 rhombomeres respectively. Each of these neuromeres is defined by a unique overlap of several genetic coding zones along the axis of the embryo. In the hindbrain and caudally to the coccyx these neuromeric units are defined by the Homeobox series of genes (Hox genes). In the forebrain, a more complex series of genes is used, such as Sonic hedgehog (Shh), Wingless (Wnt), and Engrailed (En).
Patient selection
Distinctive features of Tessier craniofacial clefts
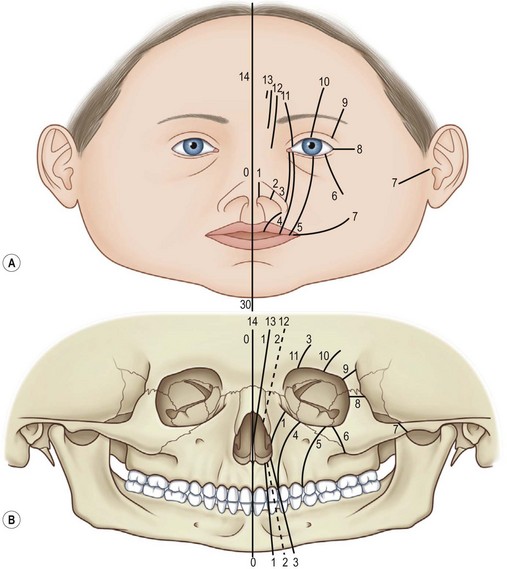
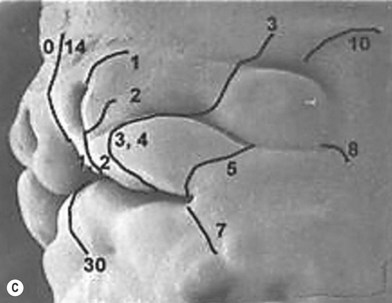
Tessier developed a classification for rare craniofacial clefts based on his anatomical and operative observations and experiences (Fig. 33.2A). The clefts are numbered from 0 to 14, follow well-defined zones of the face and orbit, and correlate with embryologic developmental maps (Fig. 33.2B). The numbered clefts relate soft-tissue clinical features to underlying bony involvement verified by operative findings and more recently preoperative 3D CT assessments.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


