Craniosynostosis, in which 1 or more cranial sutures prematurely fuse, is associated with diverse environmental and genetic factors. Whereas isolated single-suture synostosis is usually sporadic and nonfamilial, FGFR mutations account for most cases of syndromic craniosynostosis. This article reviews the etiology and various clinical manifestations of the most common isolated and syndromic forms of craniosynostosis, and provides a brief overview of genetics. Past and present surgical management approaches and techniques are examined in depth. Outcomes data in the recent literature are reviewed, and controversies in the field and promising trends in craniofacial surgery discussed.
Key points
- •
Craniofacial anomalies are common (1:2000 for isolated suture synostosis) but syndromic craniosynostosis is relatively rare.
- •
FGFR mutations underlie most syndromic craniosynostosis, whereas nonsyndromic synostosis frequently involves a variety of genetic and environmental risk factors.
- •
Single-suture synostosis is often an isolated finding, and intracranial hypertension, developmental delays, and strabismus, though less likely in isolated synostosis, are more frequent in multisuture and syndromic forms of craniosynostosis.
- •
Minimally invasive approaches are most successful when done between ages 3 and 6 months, and often require many months of molding-helmet therapy postoperatively.
- •
Surgery after age 8 to 9 months usually requires open cranial vault reconstruction whereby the skull is surgically osteotomized; bone grafts are reshaped and repositioned, then fixated in anatomically improved and often overcorrected positions.
Overview
Craniosynostosis, the premature closure of cranial sutures, may be isolated or nonsyndromic, affecting 1 in 2000 live births (typically single-sutured), or syndromic, affecting 1:30,000 to 1:100,000 live births (frequently multisutured), and primary or secondary. Multifactorial, genetic, and environmental influences may be involved. Regardless of etiology, the fused suture(s) typically cause(s) a restriction in skull growth and subsequent skull characteristics, skull base, and asymmetries and disturbances at the time of facial growth. Three theories have been proposed:
- 1.
Virchow’s theory that sutural fusion precedes other events
- 2.
Moss’s idea that primacy lies with primary skull-base growth restriction preceding synostosis
- 3.
Others have suggested that brain growth restriction is the primary factor that subsequently leads to premature fusion
It is now generally accepted that underlying mechanical force signaling pathways and cytokines mediate cranial suture patency, and premature fusion is thought to occur secondary to mutations in these genes. Secondary synostosis is evidenced in both microcephalic and overshunted individuals, and in certain hematologic disorders.
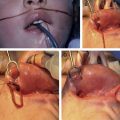
The majority or craniofacial growth and development occurs during the first year of life and growth potential, with the brain doubling in size by 1 year of life and tripling by 2 years. This rapid growth of the brain and resultant bone growth are determined by complex growth factor pathways regulated by genes that code for fibroblast growth factor receptor (FGFR) and transforming growth factor (TGF)-β, among others, and are at the core of the principles of craniofacial surgery, including the duration and timing of surgical intervention. There are clearly both environmental and genetic factors at play for isolated, single-suture, nonsyndromic forms of synostosis ( Fig. 1 ).
Overview
Craniosynostosis, the premature closure of cranial sutures, may be isolated or nonsyndromic, affecting 1 in 2000 live births (typically single-sutured), or syndromic, affecting 1:30,000 to 1:100,000 live births (frequently multisutured), and primary or secondary. Multifactorial, genetic, and environmental influences may be involved. Regardless of etiology, the fused suture(s) typically cause(s) a restriction in skull growth and subsequent skull characteristics, skull base, and asymmetries and disturbances at the time of facial growth. Three theories have been proposed:
- 1.
Virchow’s theory that sutural fusion precedes other events
- 2.
Moss’s idea that primacy lies with primary skull-base growth restriction preceding synostosis
- 3.
Others have suggested that brain growth restriction is the primary factor that subsequently leads to premature fusion
It is now generally accepted that underlying mechanical force signaling pathways and cytokines mediate cranial suture patency, and premature fusion is thought to occur secondary to mutations in these genes. Secondary synostosis is evidenced in both microcephalic and overshunted individuals, and in certain hematologic disorders.
The majority or craniofacial growth and development occurs during the first year of life and growth potential, with the brain doubling in size by 1 year of life and tripling by 2 years. This rapid growth of the brain and resultant bone growth are determined by complex growth factor pathways regulated by genes that code for fibroblast growth factor receptor (FGFR) and transforming growth factor (TGF)-β, among others, and are at the core of the principles of craniofacial surgery, including the duration and timing of surgical intervention. There are clearly both environmental and genetic factors at play for isolated, single-suture, nonsyndromic forms of synostosis ( Fig. 1 ).
Pathophysiology
Multiple environmental associations have been described, including paternal occupations such as agriculture and forestry, maternal age, exposure to tobacco smoke, and medications, including nitrofurantoin and warfarin use during pregnancy. Malpositioned fetal lie and intrauterine constraint or, at times, metabolic factors can also be associated, including mucopolysaccharidosis, mucolipidosis, rickets, and hyperthyroidosis.
Molecular genetics
A variety of mutations in transcription derived growth factors, FGFR1,2 and 3 are known to be involved in syndromic craniosynostosis ( Table 1 ).
| Syndrome | Skull Deformity | Craniofacial Abnormalities | Signs and Symptoms | Genetics | Hand and Feet Abnormalities | Incidence |
|---|---|---|---|---|---|---|
| Apert | Turribrachycephaly Acrocephaly Opened metopic suture Bitemporal bulging Bilateral coronal Bilateral lambdoid Sagittal synostosis | Orbital hypoplasia and orbital rim retrusion Hypertelorism Proptosis Downslanting palpebral fissures Cranial bony defects Short “beaked” nose Midface hypoplasia Mandibular prognatism Highly arched palate | Mental retardation Intracranial hypertension Cerebral atrophy Wide subarachnoid spaces | FGFR2 Autosomal dominant Mainly sporadic (paternal), some familial | Pansyndactylies of hands and feet | 1:65,000–100,000 live births |
| Crouzon | Acrocephaly Coronal, lambdoid, and basilar synchondroses/synostosises | Shallow orbit, orbital proptosis Hypertelorism Strabismus Beaked nose Maxillary hypoplasia Mandibular prognatism Highly arched palate Enlarged sella turcica Jugular foramina stenosis | Hydrocephalus Symptomatic chronic tonsillar herniation Syringomyelia Intracranial hypertension Optic atrophy Mental retardation | FGFR2 , FGFR3 if associated with acanthosis nigricans Autosomal dominant Sporadic and familial cases are almost equal | Usually unaffected | 1.6 per 100,000 |
| Pfeiffer |
| Orbital proptosis and stenosis Palpebral retraction Jugular foramen stenosis Midfacial hypoplasia Mandibular prognatism Highly arched palate Bifid uvula External ear duct atresia | Intracranial hypertension Hydrocephalus Sympotomatic chronic tonsillar herniation Optic atrophy Severe mental retardation (Types II and III) Transmission hearing loss Respiratory insufficiency Sleep apnea | FGFR2 FGFR1 Autosomal dominant Sporadic and familial cases are close to equal | Brachdactyly, limited variable syndactyly | 1:100,000 |
| Saethre-Chotzen | Acrocephaly Plagiocephaly Scaphocephaly Unilateral coronal, bilateral coronal, metopic, and bilateral lambdoid synostoses | Low-set frontal hairline Flat forehead Hypertelorism Eyelid ptosis Tear duct stenosis Highly arched palate Mandibular prognatism Angulated and small round ears | Mental retardation Conductive hearing loss | TWIST Gene mutation Chr 7 Autosomal dominant Mainly familial, one-third is sporadic | Partial cutaneous syndactyly, some brachydactyly | 1:25,000–1:50,000 |
| Carpenter syndrome | Sagittal and lambdoid synostosis | Downsloping palpebral fissures, low-set ears, short neck | Often short in height Obesity Mental retardation in many, normal intelligence in some | Autosomal recessive RAB23 or MEGF8 mutations | Variable polysyndactyly, clinodactyly, brachydactyly | 1:1,000,000 |
| Muenke syndrome | Unilateral or bilateral coronal synostosis | Variable hypertelorism Mild to moderate midface hypoplasia, low-set ears, associated hearing loss | Usually normal intelligence | Autosomal dominant FGFR3 mutations | Variable carpal and tarsal fusions | 1:30,000 |
Scaphocephaly
Scaphocephaly (boat-shaped head) ( Fig. 2 ) can occur without synostosis; however, it is the most common manifestation of sagittal synostosis in up to 50% of isolated synostosis. Calvarial bone growth is limited perpendicular to the affected sagittal suture, resulting in narrowing of the head transversely, and resultant brain growth anteroposteriorly leads to frontal bossing and/or occipital cupping. Shape may vary depending on the duration or timing of synostosis, whether partial or complete suture involvement, and whether other sutures are involved ( Figs. 3 and 4 ).
Trigonocephaly
Metopic synostosis may result in mild metopic ridging in the midline forehead or a combination of ridging, bitemporal narrowing, and hypotelorism, which together make the forehead appear triangular ( Fig. 5 C, D). The metopic suture fuses as early as age 3 to 6 months and, unlike other cranial sutures, normally disappears; hence, the diagnosis largely depends more on clinical shape and less on computed tomography (CT) findings.
Deformational Plagiocephaly
Meaning twisted or slanted, deformational plagiocephaly (DP) refers to flattening of the head.
The most common type of plagiocephaly is positional, namely DP. DP results when the skull is subject to pressure either in utero or, more commonly, as a result of supine positioning. Favored fetal and postnatal lie are thought to underlie DP. Congenital torticollis is associated with and likely responsible for this condition one-third of the time, leading to characteristic head tilt on the side of the affected sternocleidomastoid muscle and twisting of the neck such that babies tend to prefer sleeping on the contralateral occiput. Repositioning maneuvers and repositioning pillows work by keeping babies off the affected flat area(s), and are fairly effective in ameliorating DP when used in the first 4 to 6 months of life. Neck physiotherapy is important when congenital torticollis is present. When severe flattening persists beyond the first 5 to 6 months of life, orthotic molding bands or helmets ( Fig. 6 ) have been shown to improve cranial symmetry, especially when used before age 12 months. A recent randomized controlled study showed no differences in 5- to 6-month-old babies with moderate asymmetries treated for 6 months with or without helmets. Of note, these investigators excluded babies with the most severe cranial asymmetries.
Although DP is more commonly unilateral, bilateral DP is not uncommon and presents with brachycephaly or bilateral occipital flattening. This condition leads to a head that is short anteroposteriorly and wide transversely, which makes both the head and face look round in appearance (see Fig. 10 ).
Anterior Plagiocephaly
Unilateral coronal synostosis results in anterior plagiocephaly. The affected frontal bone is underprojected, and the contralateral frontal bone bulges anteriorly ( Fig. 7 ), which can push the orbit inferiorly, resulting in orbital asymmetry or vertical dystopia.
The superior orbital rim is elevated (Harlequin eye) ( Fig. 8 ); in severe and late presenting coronal synostosis the nasal root deviates toward the fused suture and the mandible can be similarly shifted, with additional associated ipsilateral temporal narrowing.
Posterior Plagiocephaly
Like DP, lambdoid suture synostosis results in unilateral occipital flattening, but with concomitant ipsilateral mastoid bulging and posterior inferior displacement of the ipsilateral ear. There are no frontal bony changes, whereas in DP there is concomitant ipsilateral frontal bone shift anteriorly, and in DP the ipsilateral ear is often more anterior than the unaffected side. When viewed from above these differences are described as parallelogram deformity in the case of DP and trapezoid deformity for unilateral lambdoid synostosis (see Fig. 2 ). It is worth remembering that whereas DP is common (20% to 50% of all babies are affected to some degree), lambdoid synostosis is extremely rare (1:100,000).
Brachycephaly
Although the most common cause of brachycephaly (short head) is deformational and related to supine positioning, bilateral coronal synostosis needs to be ruled out ( Fig. 9 ).
Usually a detailed history will reveal progressive flattening in positional brachycephaly, whereas syndromic brachycephaly will often present with characteristic syndromic facies and digital/pedal deformities (see Table 1 ). A noncontrasted head CT with 3-dimensional reconstruction ( Fig. 10 ) will confirm the diagnosis.
Cloverleaf skull
Cloverleaf skull is the result of fusion of all cranial sutures, except the metopic and squamosal. Because the brain is restricted in multiple planes, characteristic compensatory bulging occurs as brain and bone growth are forced across the 2 opened sutures, yielding the characteristic cloverleaf shape. Cloverleaf is perhaps the most challenging and dangerous craniosynostosis because of the severe bony growth restriction and limited intracranial volume, leading to problems with increased intracranial pressure (ICP), brain dysfunction, and hydrocephalus. Because of the severity of proptosis and orbital hypoplasia, impaired vision is common, especially in the case of extensive manipulation.
History of craniofacial surgery
History of craniofacial surgery is beyond the scope of this article Table 2 for brief overview.
| 762 bc | Homer in Iliad . Hippocrates and Galen delineate the shape |
| 16th Century | Anatomic descriptions established (Hundt, Dryander Vesalius) |
| 1851 | Virchow describes basics of abnormal and compensatory skull growth. Names condition as craniostenosis, also known as Virchow’s Law. Sear proposes title craniosynostosis in 1937 |
| 1912 | Apert and Crouzon publish syndromic craniosynostosis |
| 1890, 1892 | First reported interventions for treatment; published in Paris and San Francisco |
| 1921 | Mechner publishes first modern surgical approach |
| 1940s | Surgery available for those who present late in course of disease. Investigators propose polyethylene and tantalum to prevent suture reunion |
| 1956 | Anderson coagulates dura, based on assumption that it gives pathologic signal, but technique causes seizures. Matson applies pericraniectomy to help inhibition of bone reproduction at strip suture resection. Further development not possible without concurrent advent of modern anesthesiology and transfusion techniques. Leads to 2 deaths in 394 operations. From this point on, safety level and cosmetic outcome emerge as goals of surgery |
| 1960–1990 | Total cranial vault reconstruction with wedge craniectomies added to strip resections and Pi procedure for advanced sagittal and orbital rim. Extensive strip removal proposed and developed by Epstein, and extensive operations, new techniques, and classifications published by McComb |
| 1971 | Tessier further develops and makes safer existing surgical techniques by providing basis for safety and successful outcome |
| Today | Distraction craniotomies, endoscopic approach, and minimally invasive techniques make repertoire more colorful with obvious contribution to safety, less operative time, diminished blood loss, and shortened hospital stay |
Evaluation and diagnosis of craniosynostosis
It is important to obtain detailed history of pregnancy, birth, family, and medications or drug exposure in utero. The duration and timing of head shape is important because progressive flattening would suggest DP, with deformities that are present since birth and relatively unaffected by positioning more likely to suggest synostosis. The head should be observed from all possible views and photographic documentation obtained, fontanelles and sutures should be palpated, fronto-occipital circumference measured, and calipers used to measure cephalic index (CI) (the ratio of width/length of the skull). Careful examination of the face including orbital size, presence of asymmetry, ptosis, lagophthalmos, hypertelorism or hypotelorism, or pseudohypertelorism is warranted. Ear position, size, and any associated deformities, and facial paresis or paralysis should be noted. Full body examination is important to evaluate for findings involved in syndromic craniosynostosis, such as cardiac anomalies, respiratory difficulties as a result of mandibular or midface hypoplasia, and ocular exposure from exorbitism/proptosis. Hand/foot and finger/toe anomalies are common (see Table 1 ) with many of the syndromic craniosynostoses.
Microcephaly, venous anomalies and pressure elevation, hydrocephalus, and cerebellar tonsillar herniation all can lead to increased ICP and can present acutely, subacutely, or chronically. It is important to look for nonspecific signs and symptoms such as irritability, feeding difficulties, inconsolable crying, or insomnia as red flags for ICP elevation. A bulging fontanelle and engorged scalp veins are also often predictive. Pediatric ophthalmology evaluation to examine for strabismus and rule out papilledema is useful, and even in the absence of papilledema ICP monitoring should be considered if there is a strong suspicion for ICP elevation.
Imaging in Craniosynostosis
Skull series are sometimes obtained when evaluating for abnormal head shapes. Thumbprinting or copper-beaten appearance on plain films, representing inner table scalloping ( Fig. 11 ), is one warning of potential increased ICP.
CT scans are used to confirm a diagnosis of craniosynostosis and though not necessary for every craniofacial deformity, CT can be used with computer-aided design and modeling (CAD-CAM) for preoperative planning to create stereolithographic models, make miters, optimize osteotomies, and create custom CAD-CAM patient-specific implants designed to exactly match bony defects ( Figs. 12 and 13 ).
Some centers obtain immediate baseline postoperative CT scans following cranial vault reconstruction. Owing to the risks of ionizing radiation, the authors defer on ordering these unless the patient has neurologic deterioration; otherwise follow-up CT scans are obtained about 12 to 18 months postoperatively. For postoperative fluid collections, hematomas, or hydrocephalus, rapid magnetic resonance imaging (MRI) or lowered-radiation CT of the brain is the choice of diagnostic testing. MRI is useful for evaluating the brain for abnormalities such as Chiari malformations ( Fig. 14 ), venous anomalies, and hydrocephalus (see Table 2 ).
The concept of the craniofacial team
Because craniosynostosis is often part of a syndrome and many systems are often affected, an interdisciplinary team approach is now the standard of care for such complicated patients.
This team can include a plastic surgeon, pediatric otolaryngologist, or facial plastic surgeon trained in craniofacial surgery, a pediatric neurosurgeon, an oral-maxillofacial surgeon, a pediatric anesthesiologist and intensivist, a pedodontist orthodontist, a prosthodontist, a pediatric ophthalmologist, a psychologist, a geneticist, an audiologist, a speech pathologist, pediatrician, among others.
Treatment goals and planned outcomes
Determining which patients have or might develop functional problems as a result of increased ICP, and developmental delays, strabismus, or loss of vision as result of optic disc atrophy or exposure keratopathy is important in efforts to avoid the sequelae of these problems.
Improving outcomes from a functional standpoint is primary, but aesthetic improvements in form are not only important for cosmesis but also for improving psychosocial attitudes and perceptions, and minimizing teasing. Reconstruction aims to approximate normal size, shape, and symmetry by surgically removing synostotic bone, reconstructing the cranio-orbital asymmetries, overcorrecting when necessary, and stabilizing the reconstructed skull while minimizing immediate postoperative and/or late complications.
Preoperative planning and preparation
Physical examination is equally important in evaluating the head from all sides and from above, noting any head tilt and any associated maxillofacial, orbital, dental, or auricular asymmetries or abnormalities ( Fig. 15 ).
Several investigators have described using normative CAD-CAM templates to design fronto-orbital bandeaus and presurgical cutting guides, noting shorter intraoperative times and excellent immediate postoperative shape. The authors question long-term outcomes on such templates, as overcorrecting ( Fig. 16 ) is frequently needed for longer-lasting results.