Chapter 9
Connective Tissue Disease, Vasculitis and Related Disorders
OVERVIEW
- Many connective tissues disorders affect the skin.
- Fibrosis in connective tissue is a feature of systemic sclerosis, morphoea, CREST syndrome and lichen sclerosus.
- Lupus erythematosus may be solely cutaneous or cause severe systemic disease.
- Dermatomyositis results in characteristic skin involvement and muscle weakness, it may be a marker of internal malignancy.
- Lichen planus is a common chronic inflammatory condition of the skin of unknown cause that can affect mouth, eyes and ears.
- Vasculitis results from changes in capillaries and arterioles and may involve internal organs in addition to the skin.
- Causes of cutaneous vasculitis include inflammatory, viral and haematological conditions. A wide range of investigations may be indicated.
Introduction
The skin is a dynamic interface of trafficking immune cells that may remain localised and respond to nearby stimuli, or migrate through the skin in response to more distant triggers. The skin has been called ‘the immunological battleground of the body’ and immune cells involved in inflammatory reactions may be part of a local immune reaction or migrate to the skin as a result of antigenic stimuli at distant sites. Malfunction of the sophisticated human immune system may result in the body attacking its own tissues, that is, failure to distinguish ‘self’ from ‘non-self’. These autoimmune responses may develop against a tissue in a specific organ such as the thyroid gland, or to tissues within and between organs resulting in connective tissue diseases.
Connective tissue disease
Connective tissue disease can be difficult to define but encompasses disorders that involve tissues connecting and surrounding organs.
Connective tissues include the extracellular matrix and support proteins such as collagen and elastin. Acquired disorders of connective tissue are thought to have an autoimmune basis, many of which have distinctive clinical features and patterns in laboratory investigations. However, at times classification may not be easy. What triggers dysregulation of the immune system is usually unknown; however, some recognised factors include sunlight, infections and medication. Patients may have an underlying hereditary susceptibility to develop autoimmune diseases, marked by specific HLA (human lymphocyte antigen) types in some cases.
In autoimmune disorders immune cells may be attracted to particular targets within the skin locally (pemphigus and pemphigoid—Chapter 8) or accumulate at sites of connective tissues in multiple organs (systemic lupus erythematosus (SLE) and dermatomyositis). Once at their destination these immune cells trigger a cascade of chemical messages leading to inflammation.
The possibility of an underlying connective tissue disorder should be considered if a patient complains of any combination of symptoms including cutaneous lesions (especially face and fingers), joint pains, muscle aches, malaise, weakness, photosensitivity, Raynaud’s phenomenon and alopecia. Linking the clinical symptoms and signs with the most appropriate investigations in order to arrive at a unifying diagnosis is a challenge to even the most experienced medical practitioner (Box 9.1).
Box 9.1 Investigations might include the following
- Full blood count (FBC)
- Antinuclear antibodies (ANAs)
- Extractable nuclear antibodies (ENA), (Ro, La)
- Erythrocyte sedimentation rate (ESR)
- Renal and liver function
- ANCA (antineutrophil cytoplasmic antibodies)
- Hepatitis serology
- Streptococcal serology (ASOT)
- Rheumatoid factor
- Angiotensin-converting enzyme (ACE)
- Antiphospholipid antibodies
- Coagulation screen, lupus anticoagulant
- Anticardiolipin antibodies
- Factor V Leiden, antithrombin III, proteins S and C
- Urine dipstick and microscopy
- Blood pressure
- Chest X ray
Vasculitis
Complex reactions occurring specifically in the capillaries and arterioles of the skin may lead to cutaneous erythema (redness). The erythema may be macular or papular and may be transient or last for weeks. Blood vessels can become leaky, leading to pouring out (extravasation) of red blood cells into the tissue with or without inflammation of the blood vessel walls. Inflammation of blood vessel walls is called vasculitis and may involve arteries and/or veins. Vasculitis can also lead to stenosis, occlusion and ischaemia.
Symptoms can include pain in the skin, general malaise, fever, abdominal pain and arthropathy. Clinically, patients have a non-blanching skin eruption that is most commonly seen on the lower limbs (Figure 9.1). Individual skin lesions may be macular or palpable purpura, blistering, ulcerated and necrotic (Figure 9.2). Vasculitis confined to the skin can be painful and unpleasant, but systemic vasculitis may be life threatening.
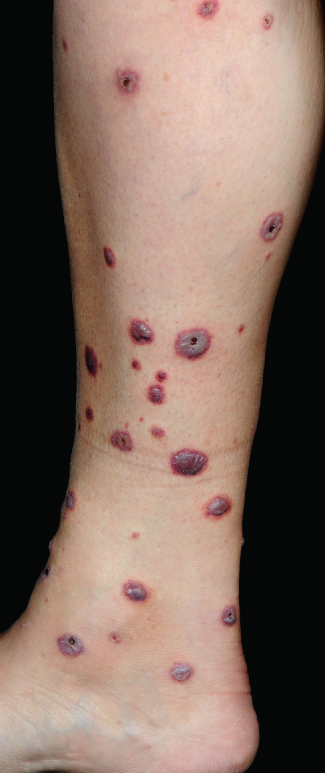
Figure 9.1 Vasculitis.
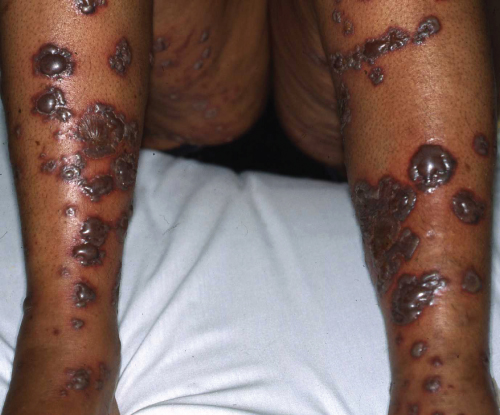
Figure 9.2 Bullous vasculitis with necrosis.
There are numerous possible underlying causes of vasculitis including infections, medications, connective tissue disease, underlying malignancy, vascular/coagulopathy disorders, inflammatory bowel disease and sarcoidosis (Box 9.1. indicates appropriate investigations in patients with a vasculitis of unknown cause).
The pathophysiology of vasculitis is complex and poorly characterised but is thought to be antibody or immune complex mediated. Blood vessel endothelial lining cells become damaged as a result of immune complex deposition, antibody targeting and consequent inflammatory cascades. Inflammation involves activation of complement and the release of inflammatory mediators resulting in vasodilatation and polymorph accumulation. The resultant leakage and occlusion of blood vessels leads to ischemia.
Confirmation of cutaneous vasculitis from a skin biopsy for histology and immunofluorescence (IMF) can be helpful but is not usually diagnostic of the underlying cause (Box 9.2). However, in Henoch–Schönlein purpura the IMF from the skin biopsy usually shows IgA deposition.
Box 9.2 Possible causes of cutaneous vasculitis
- Drug hypersensitivity
- Hepatitis
- Endocarditis
- Inflammatory bowel disease
- Connective tissue disease
- Coagulopathies
- Behçet’s syndrome
- Kawasaki disease
- Sarcoidosis
Polyarteritis nodosa (PAN)
PAN is a systemic vasculitis of small- to medium-sized arterioles that most commonly affects the skin and joints. Immune complexes mediate the disease, activating the complement cascade leading to inflammatory damage to vessels. The sites of blood vessel bifurcation are commonly affected and this leads to micro-aneurysm formation with resultant occlusion and haemorrhage. ANCA may be positive. Patients present with general malaise, fever, weight loss, weakness, arthralgia, neuropathies and skin lesions. Of the patients, 60% develop renal involvement, which may lead to renal failure. Cutaneous manifestations may include a subtle lacy/mottled pattern (livedo reticularis), purpura, tender subcutaneous nodules, ulceration and necrosis, particularly on the lower limbs. Investigations may include angiography and tissue biopsy (skin, sural nerve or muscle). Management relies on oral steroids with the addition of cyclophosphamide in severe cases.
Henoch–Schönlein purpura
This usually occurs in children (75% of cases) or young adults (M > F); the aetiology is unknown, but up to 50% of patients have preceding upper respiratory tract symptoms and a positive antistreptolysin O titre (ASOT). The skin, kidneys (IgA nephropathy), GI tract and joints are mainly affected. IgA, complement and immune complexes are deposited in small vessels (arterioles, capillaries, venules), leading to systemic vasculitis. HSP is characterised by a vasculitic rash on the buttocks and lower legs (which may associated with oedema of scrotum/hands/ears), abdominal pain and vomiting, joint pains in the knees/ankles and haematuria. Skin/renal biopsy may demonstrate deposition of IgA on immunofluorescence, which can support the diagnosis. Treatment is mainly supportive and most patients recover within weeks. Occasionally, the condition can persist and systemic corticosteroids have been used to treat skin, gastrointestinal and arthritis symptoms but steroids have not been shown to prevent or treat renal disease.
Management of cutaneous vasculitis
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


