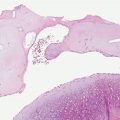
Figure 6-1 Ichthyosis vulgaris. Hyperkeratosis with a diminished and focally absent granular cell layer (original magnification ×100).
Pathogenesis. A defect in the synthesis of filaggrin, which is the major component of the granular layer, is responsible for the characteristic reduction of this layer on histopathology. Ultrastructurally, keratohyaline granules appear small and crumbly or spongy, which is evidence of defective synthesis. Defective profilaggrin expression in IV may be a result of selectively impaired posttranscriptional control (3). In addition, filaggrin links proteins of the corneocyte envelope. Dysfunctional filaggrin leads to an impaired skin barrier with increased penetration of allergens, which may predispose to epicutaneous sensitization and the development of atopic disorders, including atopic dermatitis and asthma. Studies suggest that approximately 25% to 50% of patients with atopic dermatitis and secondary asthma harbor filaggrin mutations, approximately 40 of which have been identified as pathogenic (4).
Differential Diagnosis. Although the noninflamed but dry skin of patients with atopic dermatitis clinically resembles IV, on histologic examination, atopic dermatitis more commonly demonstrates epidermal hyperplasia, patchy parakeratosis, and slight hypergranulosis.
X-Linked Recessive Ichthyosis (XLRI)
Clinical Summary. X-linked recessive ichthyosis (XLRI) is caused by a mutation in the ARSC1 gene encoding steroid sulfatase, which in nearly 90% of cases results from gene deletion. It rarely presents at birth and most frequently develops in the first few months of life. Males are invariably affected. Female carriers do not tend to manifest the clinical cutaneous phenotype because the gene is located at the distal tip of the X chromosome, which typically escapes random X-inactivation. As a result, both genes are expressed in each cell, providing sufficient enzyme in spite of the mutated allele that the skin remains largely unaffected; however, female carriers who are pregnant often have prolonged labor and may fail to progress due to insufficient placental steroid sulfatase. Affected females with Turner syndrome or those with homozygous mutations in the ARSC1 gene have been described and those so affected have more typical features of the disorder (5). Patients tend to develop plate-like brown scale diffusely on the trunk and extremities, with sparing of the flexural areas (as in ichthyosis vulgaris) and the palms, soles, and central face. The preauricular regions and neck are often involved. Asymptomatic corneal opacities have been described in 25% to 50% of patients, and can be seen in unaffected carrier females as well (6). Boys may also be affected with isolated cryptorchidism or hypogonadism (7). A contiguous gene syndrome has been observed in patients with clinical findings of XLRI and Kallman syndrome (hypogonadotropic hypogonadism associated with mental retardation and anosmia) (8). In the uncommon instances where extracutaneous features are identified, they are considered examples of syndromic ichthyosis.
Histopathology. Hyperkeratosis is present along with a granular layer that is normal or slightly thickened, in contrast to IV. The epidermis may be slightly acanthotic as well (9).
Pathogenesis. Steroid sulfatase is concentrated in lamellar bodies and degrades cholesterol sulfate, which provides the cholesterol “mortar” between “brick” corneocytes in the cornified envelope. Cholesterol sulfate also functions as a serine protease inhibitor in the epidermis and is required for orderly corneocyte degradation and normal desquamation. Increased cholesterol sulfate leads to persistent cell cohesion with delayed dissolution of desmosomes in the stratum spinosum, leading to retention hyperkeratosis (10,11). This was first recognized in skin fibroblasts but was found subsequently also in the entire epidermis and in leukocytes (10,12).
Keratinopathic Ichthyosis
Keratinopathic ichthyosis is the updated terminology for ichthyoses resulting from mutations in keratin genes, which include epidermolytic ichthyosis (the most common subtype and previously referred to as bullous congenital ichthyosiform erythroderma (BCIE), ichthyosis en confetti, and superficial epidermolytic ichthyosis (previously called ichthyosis bullosa of Siemens).
Epidermolytic Ichthyosis
Clinical Summary. Epidermolytic ichthyosis (EI) is caused by mutations in the keratin 1 (KRT1) or keratin 10 (KRT10) genes and is typically inherited in an autosomal dominant fashion, although recessive forms have been reported (1). Infants show generalized tender erythema from the time of birth, with the development of superficial bullae and erosions that can be confused with staphylococcal scalded skin syndrome. Vesicles, bullae, and erosions typically resolve within the first few years, to be replaced by thick brown, verrucous scaling that predominates over time (Fig. 6-2). The flexural surfaces show marked involvement with furrowed hyperkeratosis, particularly in flexural areas, leading to the appearance of “corrugated cardboard.” Patients with severe disease are commonly colonized with bacteria and may exhibit a characteristic odor.
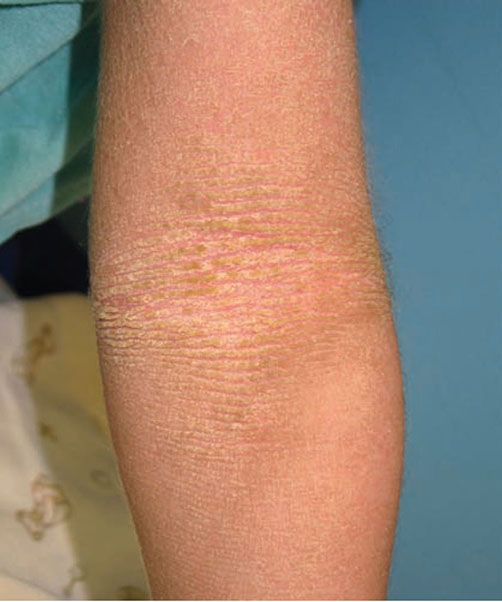
Figure 6-2 Epidermolytic ichthyosis. Linear scaling reminiscent of “corrugated cardboard.” (Image courtesy of CHOP dermatology.)
Histopathology. Epidermolytic hyperkeratosis (EHK) is a characteristic histologic finding in the epidermis (Fig. 6-3). There are variously sized clear spaces around the nuclei in the upper stratum spinosum and in the stratum granulosum. Peripheral to the clear spaces, the cells show indistinct boundaries formed by lightly staining material or by keratohyaline granules. A markedly thickened granular layer containing an increased number of irregularly-shaped keratohyaline granules and compact hyperkeratosis is observed (13). When bullae form, they arise intraepidermally through separation of edematous cells from one another. The upper dermis shows a moderately severe, chronic inflammatory infiltrate (14).
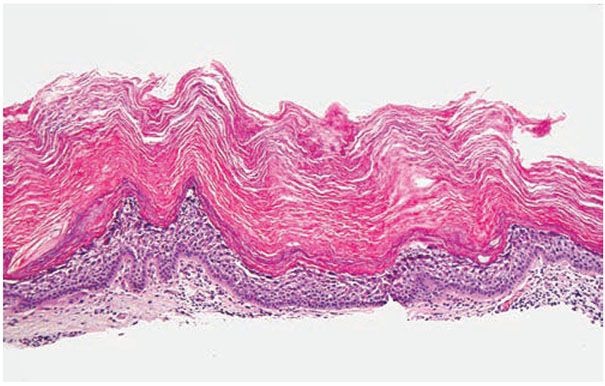
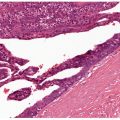
Figure 6-3 Epidermolytic hyperkeratosis. There is vacuolization of the upper and mid-spinus layer, with hyperkeratosis and large keratohyaline granules in the vacuolated expanded granular cell layer (original magnification ×100).
Pathogenesis. Mutations are predominantly point mutations that occur in the carboxy terminal of the rod domain of keratin 1 and the amino terminal of the rod domain of keratin 10, producing an abnormal keratin network that leads to blistering and skin fragility (15). The essential electron microscopic features are excessive production of tonofilaments and premature formation of keratohyaline granules. At the periphery of the cells, numerous keratohyaline granules are embedded in thick shells of irregularly clumped tonofilaments. The desmosomes appear normal, but the tonofilament–desmosomal interaction is disturbed, so many desmosomes are attached to only one keratinocyte instead of connecting two neighboring keratinocytes, with resultant clinically observed blister formation and sometimes histologic features of acantholysis.
Differential Diagnosis. Although EHK is found in all cases of EI, it can also be seen as an isolated histologic finding in other benign dermatoses and neoplasms, including epidermolytic palmoplantar keratoderma (detailed below), solitary epidermolytic acanthoma, disseminated epidermolytic acanthoma, and an epidermal nevus, usually of the systematized type. In the latter case, it represents a mosaic finding where only affected skin (but not normal surrounding skin) contains a mutation in the keratin 1 or 10 genes (16). Patients with extensive epidermal nevi exhibiting EHK may possess germ line mutations, which may place them at risk for transmitting germ line mutations to their offspring, resulting in generalized EI (17).
Ichthyosis En (with) Confetti
Clinical Summary. Ichthyosis with confetti is an autosomal dominant disorder also caused by mutations in KRT1 or KRT10. Affected patients are born with exfoliative erythroderma and palmoplantar keratoderma, which over many years becomes peppered with numerous pale “confetti-like” islands of normal skin during childhood (18).
Histopathology. Histology of the erythrodermic skin shows epidermal acanthosis with disordered differentiation above the basal layer, perinuclear vacuolization, hypogranulosis, and hyperkeratosis with retained nuclei in the stratum corneum (19).
Pathogenesis. Ichthyosis with confetti has been shown to result from revertant mosaicism, where islands of normal intervening skin appear due to loss of heterozygosity in the KRT10 gene via mitotic recombination (19). This form of “natural gene therapy” results from genetic events in a single somatic cell, causing loss of the disease-causing phenotype, followed by expansion of the reverted cell. Revertant mosaicism has been reported in other skin diseases, including epidermolysis bullosa as well as in primary immunodeficiencies and muscular dystrophy (20).
Superficial Ichthyosis
Clinical Summary. Superficial ichthyosis, previously called ichthyosis bullosa of Siemens, is caused by mutations in the KRT2e gene, and is marked by more superficial blistering than EI, which is consistent with localization of KRT2 in the upper spinous and granular layers. Intact bullae can occur, but denuded peeling skin more commonly predominates. Hyperkeratosis of the palms and soles is rare.
Histopathology. Skin biopsies are similar to EI with EHK in the upper spinous and granular layers. The diagnosis is often a clinical one, with genetic confirmation of mutations in KRT2 (21).
Autosomal Recessive Congenital Ichthyosis
Clinical Summary. ARCI is a collection of various disorders that includes a range of phenotypes including harlequin ichthyosis (HI), lamellar ichthyosis (LI), and nonbullous congenital ichthyosiform erythroderma (CIE), but some patients may have overlapping features. The diagnosis is exclusively a clinical one, although biopsy can be helpful to exclude other diagnoses. HI is the most severe ARCI phenotype, and patients are born with fissured ichthyosiform polygonal plaques that mimic the costume of a harlequin (Fig. 6-4). Patients with the CIE phenotype demonstrate fine white scales with fairly pronounced erythroderma that may improve with age. The LI phenotype is characterized by large, plate-like “fish” scales, although there is a spectrum of severity ranging from mild disease to more severe involvement with ectropion (eversion of the eyelid resulting in exposure of the conjunctiva), eclabium (eversion of the lips), and scarring alopecia (Fig. 6-5). In all forms, the flexural surfaces and the palms and soles are involved.

Figure 6-4 Harlequin ichthyosis phenotype of ARCI. Polygonal fissured plaques on the trunk of this neonate with a confirmed ABCA12 mutation. (Image courtesy of CHOP dermatology.)
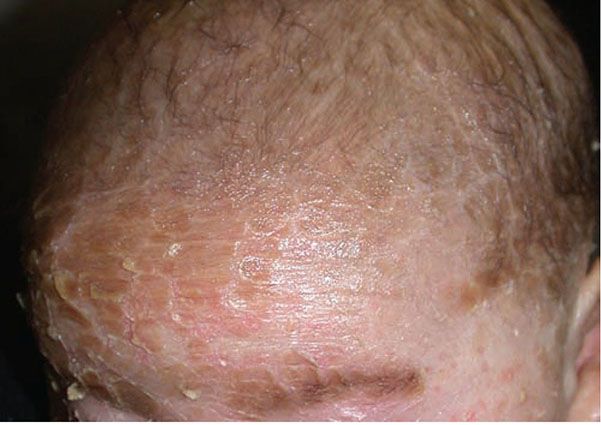
Figure 6-5 Lamellar ichthyosis phenotype of ARCI. Plate-like scaling on the forehead of a patient with confirmed TGM-1 mutation. (Image courtesy of CHOP dermatology.)
Pathogenesis. Six genes have been implicated in ARCI. Most patients with the lamellar phenotype have mutations in the gene encoding transglutaminase 1 (TGM1), which cross-links proteins in the cornified envelope (22). HI results from nonsense mutations in the ABCA12 transporter gene, leading to deficiency of proteases in the stratum corneum, dysregulated lipid transport with abnormal lamellar bodies, and premature differentiation of keratinocytes. Massive orthokeratosis results (23). Missense mutations in ABCA12 produce milder LI or CIE phenotypes due to the presence of some functioning protein (24). Mutations in ALOX12B (12(R)-lipoxygenase, ALOXE3 (lipoxygenase-3), CYP4F22 (cytochrome p450 subunit), and NIPAL4 (ichthyin) also lead to ARCI through disruption of lipid synthesis and/or processing of filaggrin (25–28).
Histopathology. The histologic findings are nonspecific and include orthokeratosis with follicular plugging. Typically, histopathology of CIE shows mild thickening of the stratum corneum with foci of parakeratosis, while LI demonstrates a markedly thickened stratum corneum without areas of parakeratosis. On electron microscopy, HI demonstrates absent lamellar granules in the stratum granulosum (29).
Syndromic Ichthyoses
Clinical Summary. There is an expanding list of syndromes that combine ichthyosis with neuroectodermal and mesodermal defects. Most of these syndromes demonstrate nonspecific orthokeratosis with the exception of Refsum disease, for which the histopathologic skin findings are specific and diagnostic, and CHILD syndrome (see below).
Sjögren–Larsson syndrome is an autosomal recessive disorder characterized by erythroderma early in life, with fine white scaling in flexural areas with mental retardation and spastic paresis. Most patients are photophobic but “retinal glistening dots” are not always present in childhood. Collodion presentation is rare. With age, these patients develop generalized lamellar thickening, which is most pronounced on the neck and trunk, sparing the face. Pruritus is invariably present and can be severe (30). The disorder is due to mutations in the fatty aldehyde dehydrogenase gene (FALDH), which converts fatty alcohol to fatty acid (31).
Conradi–Hunermann syndrome is an X-linked dominant form of chondrodysplasia punctata, which is characterized by punctate epiphyseal calcifications, asymmetric skeletal abnormalities, ichthyosiform areas in a blaschkoid distribution (termed ptychotropism), and cataracts. X-chromosomal inactivation in girls produces the blaschkoid skin findings and asymmetric bone involvement, and the underlying defect occurs in the pathway of cholesterol synthesis and metabolism. Mutations have been found in the emopamil-binding protein (EBP) gene, which encodes 3β-hydroxysteroid delta 8, delta 7 sterol isomerase (32). Some forms are associated with peroxisome abnormalities (32).
Netherton syndrome (NS) is an autosomal recessive condition marked by the triad of ichthyosis, atopic diathesis, and hair shaft deformities, most commonly trichorrhexis invaginata. Most patients present in the newborn period with an exfoliative erythroderma and failure to thrive, and they are at high risk for sepsis and electrolyte abnormalities due to dehydration. The classic skin finding in NS is ichthyosis linearis circumflexa, which is not typically seen until early childhood and manifests as extensive migratory polycyclic lesions of erythema and scaling. At their periphery, some of the areas show a distinctive “double-edged” scale (33). Trichorrhexis invaginata (so-called “bamboo-hair”) is the classic hair shaft deformity, giving the hair the appearance of bamboo. This finding is thought to represent a defect involving the inner root sheath (34). Patients typically have elevated eosinophils and IgE with severe atopy including eczema, asthma, food allergies, and anaphylaxis (35). The skin barrier dysfunction in these patients results in a high risk for percutaneous absorption of topically applied medicaments.
Histopathology. The areas of erythema and scaling show nonspecific changes with some resemblance to psoriasis, such as elongation of the rete ridges and hyperkeratosis, as well as parakeratosis. The double-edged scale frequently manifests as spongiosis in the upper stratum spinosum, resulting in multilocular vesicles or vesiculopustules (36).
Pathogenesis. NS is caused by mutations in the gene SPINK5, which encodes LEKTI (Lymphoepithelial Kazal-Type Inhibitor), a serine protease inhibitor. Deficiency of LEKTI causes epidermal protease hyperactivity, leading to desmosomal degradation, detachment of the stratum corneum, and activation of TH2 cytokines, in turn leading to atopy (33,37).
Trichothiodystriphy is otherwise known as IBIDS syndrome due to characteristic features of Ichthyosis, Brittle hair, Impaired intelligence, Decreased fertility, and Short stature. Some patients also demonstrate photosensitivity (which is referred to as PIBIDS syndrome). Patients with these syndromes have sulfur-deficient, sparse hair, which demonstrates a characteristic “tiger tail” appearance under polarized microscopy (38).
Refsum disease is an autosomal recessive disorder characterized by generalized ichthyosis, cerebellar ataxia, progressive paresis of the extremities, and retinitis pigmentosa, which typically manifests in late childhood or adolescence. Patients typically present with neurologic manifestations, and a delay in diagnosis can lead to irreversible damage.
Histopathology. The skin shows hyperkeratosis, hypergranulosis, and acanthosis. Pathognomonic findings occur in the basal and suprabasal cells of the epidermis, which demonstrate variably sized vacuoles that contain lipid accumulations (39).
Pathogenesis. Refsum disease is caused by mutations in two genes: the PAHX gene, which encodes the peroxisomal enzyme phytanol-CoA hydroxylase, and the PEX7 gene, which encodes the PTS2 (peroxisomal targeting signal 2) receptor, both of which lead to the accumulation of phytanic acid (40,41). Treatment is avoidance of phytanic acid-containing foods.
CHILD syndrome is an X-linked dominant disease characterized by Congenital Hemidysplasia with Ichthyosiform erythroderma and Limb Defects. Its hallmark is a unilateral ichthyosiform erythroderma that does not cross the midline with ipsilateral hypoplasia of the affected limb (Fig. 6-6). Nearly all published cases have been female, and it is presumed to be lethal in the hemizygote male fetus. The disorder is due to mutations in the NSHDL gene, which encodes the enzyme 3β-hydroxysteroid dehydrogenase that converts lanosterol to cholesterol (42). Ultrastructural analysis of affected skin has demonstrated both cholesterol depletion and accumulation of toxic metabolites. Application of topical lovastatin/cholesterol compound (but not cholesterol alone) has resulted in virtual clearance of affected skin, with normalization of histologic and ultrastructural skin, presumably due to therapy that not only replenished deficient end-product but also prevented generation of toxic metabolites (43).
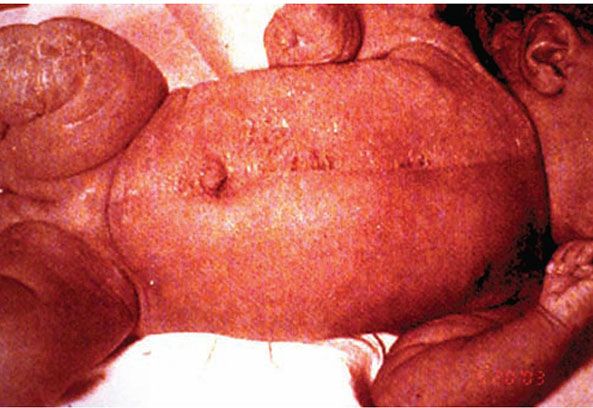
Figure 6-6 CHILD syndrome. Note unilateral erythema, scaling, and ipsilateral limb hypoplasia.
Histopathology. The epidermis is acanthotic with alternating layers of orthokeratosis and parakeratosis in the stratum corneum. Numerous lipid-laden vesicles can be noted in the lower stratum corneum, with elongation of the rete ridges and an inflammatory infiltrate (44). Histologic features of verruciform xanthoma (VX) can be seen in the areas of ichthyosiform erythroderma and isolated VX can also be seen in CHILD syndrome. Histologically, VX is characterized by infiltration of papillary dermal tips with foamy macrophages (45).
Principles of Management. The current standard for treating all forms of ichthyosis includes keratolytics and emollients containing urea, lactic or glycolic acid, or propylene glycol to restore the epidermal barrier (46,47). Salicylic acid-based products can also be effective, but they should be used with great caution in these disorders where percutaneous absorption can be significant and can lead to potential toxicity. There is emerging evidence for use of topical N-acetyl cysteine, a glutathione precursor, which serves various functions as a potent antioxidant and may also have effects on improving keratinocyte differentiation (47). Topical steroids are often ineffective as there is minimal epidermal hyperproliferation or inflammation. CHILD syndrome has responded to topical application of 2% lovastatin/2% cholesterol compound to affected skin to bypass the defective cholesterol synthesis pathway and reduce toxic metabolites associated with NSDHL deficiency (48).
OTHER DISORDERS OF KERATINOCYTE DIFFERENTIATION
Erythrokeratoderma Variabilis and Progressive Symmetric Erythrokeratoderma
Clinical Summary. A rare, autosomal dominant, inherited disorder, erythrokeratoderma variabilis (EKV), typically starts in infancy and is characterized by two striking morphologies. First, areas of erythema expand centrifugally and coalesce into circinate figures. These lesions fluctuate over days to weeks in their configuration and extent and thus are “variable.” Second, persistent yellow to brown hyperkeratotic plaques develop both within the areas of erythema and in areas of apparently normal skin. The trunk, buttocks, and limbs are typically affected with relative sparing of the face (49). Progressive symmetric erythrokeratoderma (PSEK) is a variant of EKV characterized by fixed, symmetric hyperkeratotic plaques involving the extensor surfaces, the face and buttocks. The disorder is inherited with variable penetrance and is best distinguished from EKV by the facial involvement and absence of migratory erythema, although overlap phenotypes have been reported (50,51).
Histopathology. The changes are nonspecific in both EKV and PSEK. The hyperkeratotic plaques consist of hyperkeratosis with moderate papillomatosis and acanthosis. The granular layer appears normal, being two to three cell layers thick (49).
Pathogenesis. EKV is caused by mutations I GJB3 encoding connexin 31 (52) and GJB4 encoding connexin 30.3 (53,54). Labeling with tritiated thymidine shows a normal rate of proliferation, and it is likely that the hyperkeratosis is due to retention of corneocytes due to decreased shedding of horny cells. The genetics of PSEK are still poorly understood, although missense mutations in connexin 30.3 and the loricrin gene have previously been reported in some patients (55–57).
Principles of Management. Patients tend to improve with age and during warmer months, and many are asymptomatic. Liberal use of emollients is a mainstay of treatment. Systemic retinoids have been used in some cases with improvement.
Palmoplantar Keratodermas
Clinical Summary. The palmoplantar keratodermas (PPKs) (Table 6-1) represent a heterogeneous group of diseases that share the clinical characteristic of prominent thickening of palmoplantar skin. The molecular pathogenesis of the inherited forms are linked to abnormalities in intracellular structural proteins (keratins), cornified envelope proteins (loricrin, transglutaminase), cell–cell connections (desmoglein, desmoplakin, plakophilin, connexins), and enzymatic signaling transduction (cathepsins) (58,59). They are typically classified as diffuse or focal. The major forms of diffuse PPK include
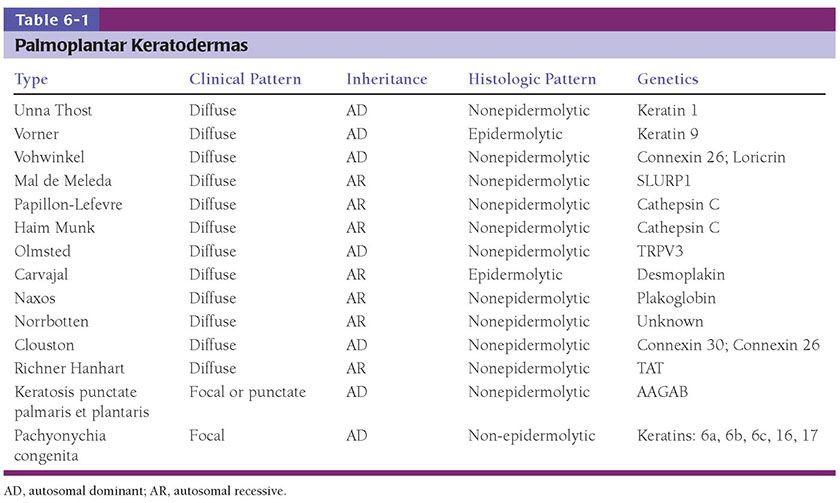
1. Diffuse nonepidermolytic palmoplantar keratoderma, otherwise known as Unna Thost type, is an autosomal dominant disorder characterized by diffuse hyperkeratosis of the palms and soles (Fig. 6-7). It is caused by mutations in the gene encoding keratin 1 (60).
Figure 6-7 Keratosis palmeris et plantaris of Unna-Thost. Thickened palms and soles of the father, whose child has similar findings.
2. Diffuse epidermolytic palmoplantar keratoderma, otherwise known as the Vorner type, is clinically indistinguishable from the Unna-Thost type but histologically demonstrates epidermolytic hyperkeratosis. This disorder is inherited in an autosomal dominant fashion and is often associated with hyperhidrosis and secondary bacterial infection. This variant has been associated with mutations in keratin type 9 localized within the keratin gene cluster on chromosome 17q 12-q21 (58,61).
3. Vohwinkel syndrome is an autosomal dominant disorder associated with sensorineural deafness, severe mutilating PPK with a characteristic honeycomb pattern, and constrictions called pseudoainhum that may lead to auto-amputation. Hyperkeratotic starfish-like plaques are commonly observed on the knees, elbows, and dorsal hands. It is caused by mutations in the GJB2 gene, which encodes connexin 26 (62,63). A variant of Vohwinkel syndrome with prominent PPK and ichthyosis, but normal hearing is caused by mutations in the gene that encodes loricrin (64).
4. Mal de Meleda is an autosomal recessive PPK with diffuse inflammatory keratoderma of the palms and soles that extends to involve the dorsal hands, feet, ankles, and wrists (referred to as transgradiens involvement). Flexion contractures may occur when keratoderma overlies joints, and autoamputation is a rare complication. The disorder is caused by mutations in SLURP1, a secreted epidermal neuromodulator that regulates epidermal homeostasis and inhibits macrophage-induced TNF-α release, accounting for its inflammatory and hyperproliferative phenotype (65).
5. Like Mal de Meleda, Papillon–Lefèvre syndrome is a transgradiens PPK with hyperkeratosis of the palms and soles that extends to the dorsal hands and feet. Patients often have psoriasiform plaques on the elbows and knees. Its distinguishing feature is early-onset, severe periodontitis, resulting in loss of both deciduous and permanent teeth with secondary changes in alveolar bone (66). Haim Munk syndrome (reported in individuals from a single religious isolate in Cochin, India) is a variant characterized by periodontitis, with additional findings of acroosteolysis, onychogryphosis, and arachnodactyly. Both disorders have been localized to abnormalities in cathepsin C localized to chromosome 11q14.1 and result from activation of serine proteases, leading to premature desquamation (61).
6. Olmsted syndrome (mutilating palmoplantar keratoderma with periorificial plaques) is an autosomal dominant PPK characterized by mutilating keratoderma of the palms and soles, periorificial and often intertriginous inflammatory plaques, and alopecia that begins in infancy. The disorder can be complicated by severe contractures with auto-amputation from progressive constriction. Recently, mutations have been found in the TRPV3 gene, which encodes a transient receptor potential vanilloid-3 cation channel, leading to apoptosis of keratinocytes with reactive hyperplasia of the epidermis (67).
Histopathology. The histopathology for all of the diffuse PPKs except for diffuse epidermolytic palmoplantar keratoderma is nonspecific, consisting of considerable hyperkeratosis, hypergranulosis, acanthosis, and a sparse inflammatory infiltrate of lymphocytes in the upper dermis (68). Diffuse epidermolytic palmoplantar keratoderma demonstrates an identical histology to that seen in epidermolytic hyperkeratosis. Many cells in the middle and upper stratum spinosum appear vacuolated, and scattered cavities are present as a result of ruptured cell walls, with numerous large keratohyaline granule (69,70).
The focal (or punctate) PPKs include:
Keratosis Punctata Palmaris et Plantaris
Clinical Summary. This is an autosomal dominant disorder characterized by multiple discrete keratotic plugs on the palms and soles, which typically manifests in adolescence.
Histopathology. There is massive hyperkeratosis over a sharply limited area, with depression of the underlying malpighian layer below the general level of the epidermis. There is an increase in the thickness of the granular layer. The dermis is free of inflammation (71).
Pathogenesis. At least three forms of the disorder have been described, some of which have been mapped to the AAGAB gene, which encodes an alpha-and-gamma-adaptin-binding protein p34 that serves as a chaperone for membrane trafficking (72,73).
Pachyonychia Congenita (PC)
Clinical Summary. PC refers to a collection of autosomal dominant disorders characterized by early-onset nail dystrophy followed by the development of focal painful palmoplantar keratoderma sometimes with secondary bullae formation. Other notable findings include mucosal leukokeratosis (Fig. 6-8), occasionally cysts, follicular keratoses on the elbows and knees, and natal teeth. Unlike other conditions associated with oral leukokeratoses, the mucosal changes in PC are benign and do not predispose to malignant degeneration (Fig. 6-9).
Figure 6-8 Pachyonychia congenita. Thickened nails in infant with this syndrome.
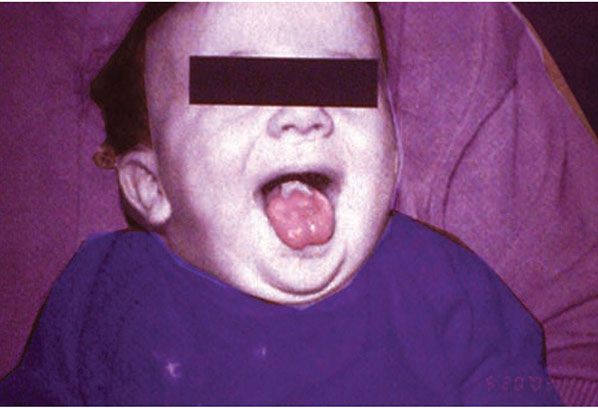
Figure 6-9 Pachyonychia congenita. Note whitened mucosa at back of tongue.
Histopathology. The nail bed shows marked hyperkeratosis. As in the normal nail bed, there is no granular layer. The blisters that may be seen beneath and around the plantar callosities arise in the upper layers of the stratum spinosum through increasing intracellular edema and vacuolization. Unlike friction blisters, they show no areas of necrosis (74). The oral lesions show thickening of the oral epithelium with extensive intracellular vacuolization, exactly as seen in white sponge nevus, and without evidence of dyskeratosis (75).
Pathogenesis. Genetic mutations have been described in the paired keratin genes 6a, 6b, 6c, 16, and 17, which are variably expressed in the nail bed, mucosa, and palmoplantar epithelium, and lead to epidermolysis with compensatory reactive hyperkeratosis. The historical terms Jadassohn–Lewandosky syndrome (PC-1) and Jackson–Lawler syndrome (PC-2) have been abandoned due to considerable overlap between subtypes in favor of a new classification scheme that categorizes disease phenotype based on specific keratin mutations. The presence of cysts, previously thought to be pathognomonic of PC-2, associated with keratin 17 mutations have now been documented in patients with keratin 6a mutations. Genotype–phenotype investigation is still ongoing through the International Pachyonychia Congenita Research Registry, but certain clinical phenotypes appear to point toward particular keratin mutations. For example, congenital nail dystrophy that involves all nails is most likely to indicate keratin 6a or 17 mutations. The concurrent presence of congenital nail dystrophy and natal teeth strongly suggests keratin 17 mutations. Childhood PPK with later development of associated features points toward keratin 16 mutations, and isolated focal nail involvement is rare but most likely to indicate underlying keratin 6c mutations (76).
PPK can also be seen in association with other unique identifying features, for example, Norrbotten recessive palmoplantar keratoderma (associated with knuckle pads). Those inherited disorders featuring PPK along with extracutaneous signs include Carvajal syndrome (dilated cardiomyopathy with epidermolytic keratoderma), Naxos disease (woolly hair, diffuse nonepidermolytic palmoplantar keratoderma, with arrythmogenic cardiomyopathy), hidrotic ectodermal dysplasia (Clouston syndrome), and Richner-Hanhart syndrome (tyrosinemia type II).
Principles of Management. Treatment of all forms of PPK includes keratolytics, soaks, and debridement as necessary, particularly for the mutilating disorders. Systemic retinoids can be helpful but should be reserved for those with severe disease and disability, given their long-term side effects.
Acrokeratoelastoidosis
Clinical Summary. Acrokeratoelastoidosis is a rare condition characterized by shiny, firm, yellow-to-translucent, sometimes umbilicated papules, which develop at the periphery of the palms and soles with extension to the dorsa of the fingers and the sides of the feet (Fig. 6-10) (77). Autosomal dominant inheritance is most common, although some cases appear to be sporadic. Most patients develop the disorder in childhood or adolescence but presentation in adulthood has been reported. Associated findings may include diffuse palmoplantar keratoderma and hyperhidrosis (78).
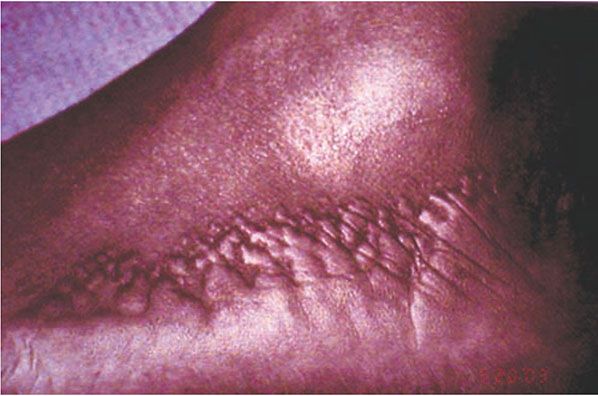
Figure 6-10 Acrokeratoelastoidosis. Firm papules on the lateral foot.
Histopathology. Elastorrhexis is the essential histologic feature of acrokeratoelastoidosis, with diminution and fragmentation of the elastic fibers, especially in the deeper portions of the papillary dermis sparing the reticular dermis (77). Some of the fragmented elastic fibers may appear thickened and tortuous (79). Additional histopathologic findings include focal hyperkeratosis, acanthosis, and hypergranulosis (80).
Pathogenesis. The pathogenesis remains speculative. There is some data to suggest that acrokeratoelastoidosis results from failed secretion of elastin from fibroblasts, rather than from degeneration of elastic fibers (81). Formation of the discrete papules may result from filaggrin aggregation (82).
Differential Diagnosis. Focal acral hyperkeratosis looks identical clinically but lacks elastorrhexis on histopathology (80). Degenerative collagenous plaques of the hands are typically concentrated on the index finger and opposing thumb in white men with chronic sun exposure and repetitive trauma from manual labor. There is no familial predisposition. Histologic changes consist of basophilic degeneration of the elastic tissue (77).
Principles of Management. Most patients are asymptomatic and require no treatment. Topical keratolytics such as urea 40% cream have been reported to be helpful to some patients.
The Porokeratoses
The term “porokeratosis” refers to a group of disorders characterized by a distinct “thread-like” peripheral hyperkeratotic ridge that histologically corresponds to the cornoid lamella, a column of parakeratosis arising from an invagination of the epidermis (Fig. 6-11). It was originally coined by Mibelli in 1893, who erroneously believed that the cornoid lamella arose from sweat pores (83).
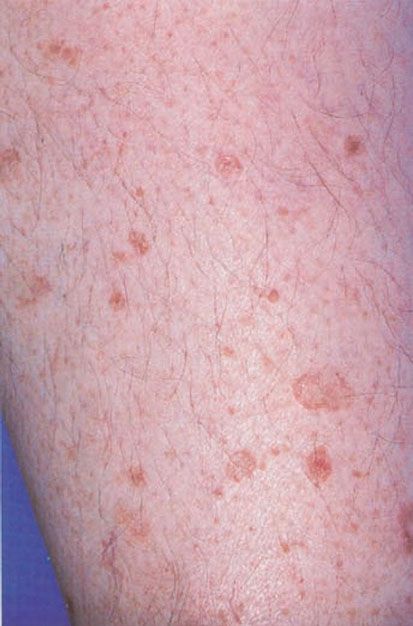
Figure 6-11 Disseminated actinic porokeratosis. Numerous pink-brown plaques on the leg, with a characteristic raised peripheral rim. (Photograph by William K. Witmer.)
Clinical Summary. The porokeratoses are speculated to arise from a local clonal proliferation of abnormal keratinocytes, although the genetics are still incompletely understood. Five different forms have been described (84).
One or more annular, hyperkeratotic plaques that develop in childhood and subsequently enlarge from the periphery over years characterize classic porokeratosis of Mibelli. Lesions initiate as hyperkeratotic papules that can enlarge to several centimeters in diameter. The characteristic keratotic border has been likened to the “Great Wall of China” and correlates to the cornoid lamella on histopathology. Rarely, numerous lesions may develop, and these are typically unilateral. Boys are more commonly affected. Transmission may be autosomal dominant with incomplete penetrance, or through a sporadic event (85).
Disseminated superficial actinic porokeratosis (DSAP), the most common of the porokeratoses, is an autosomal dominant disorder with incomplete penetrance that typically manifests in the third or fourth decade of life (86). Multiple (>50) atrophic circinate plaques with a thread-like border occur most commonly on sun-exposed surfaces, typically the lower legs and arms, and may be exacerbated by exposure to sun (Fig. 6-11). However, the term actinic cannot be applied to all cases. The main risk factors for DSAP include sun exposure, genetic susceptibility, and immunosuppression, presumably because of reduced immune surveillance (84). Interestingly, immunosuppressed patients with DSAP do not appear at greater risk for malignant transformation than other patients, in spite of a higher risk of de novo skin cancers (87). Mutations in the mevalonate kinase (MVK) gene have been found in some patients, although the function of this enzyme in DSAP pathogenesis is unclear (88).
Linear porokeratosis may involve only a segment of the body or may be generalized, but typically follows Blaschko’s lines (Fig. 6-12). Presentation in infancy or childhood is most common. It likely represents a form of type II mosaicism, reflecting loss of heterozygosity in early development with proliferation of a single-cell clone (89).
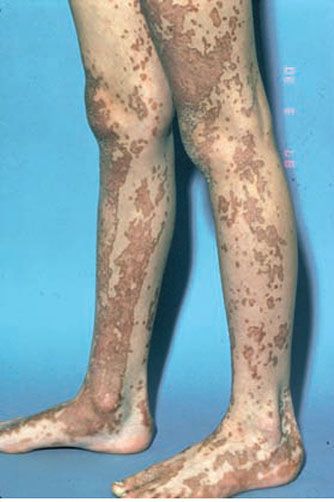
Figure 6-12 Linear porokeratosis. Linear hyperkeratosis along the feet and legs.
Punctate porokeratosis, with onset in childhood or adolescence, is characterized by numerous punctate 1- to 2-mm seed-like keratotic plugs limited to the palms and soles, without tendency to centrifugal enlargement. They may be moderately tender to pressure (90).
Porokeratosis plantaris, palmaris, et disseminata is an autosomal dominant variant of punctate porokeratosis characterized by keratotic papules on the palms and soles in childhood or early adolescence with subsequent generalization to other areas of the body. Both sun-exposed and sun-protected skin may be involved. Males are more commonly affected, but the genetic cause remains unknown (91,92).
The development of nonmelanoma skin cancers (predominantly squamous cell carcinoma or Bowen disease) within lesions of porokeratosis is well documented, with increased p53 expression found in some cases (93,94). A review of 281 individuals with porokeratosis from 1964 to 1994 found the highest incidence of skin carcinoma development in patients with linear porokeratosis (19%), followed by porokeratosis palmaris and plantaris (9.5%), porokeratosis of Mibelli (7.6%), DSAP (3.4%), and punctate porokeratosis (0%) (87). None of the patients who developed cutaneous malignancies were iatrogenically immunosuppressed, although two patients had congenital genodermatoses (Werner and Bloom syndromes).
Histopathology. It is important that the biopsy specimen include the peripheral, raised, hyperkeratotic ridge. On histologic examination, the ridge correlates to a keratin-filled invagination of the epidermis. In classic porokeratosis of Mibelli, the invagination extends deeply downward at an angle, the apex of which points away from the central portion of the lesion. In the center of this keratin-filled invagination rises a parakeratotic column, the cornoid lamella, representing the most characteristic feature of porokeratosis (Fig. 6-13) (83). Within the parakeratotic column, the cells appear homogeneous and possess pyknotic nuclei. In the epidermis beneath the parakeratotic column, the keratinocytes are irregularly arranged and have pyknotic nuclei with perinuclear edema. Dyskeratotic keratinocytes can be seen in the epithelium at the base of the cornoid lamella. In the upper stratum spinosum, some cells possess an eosinophilic cytoplasm as a result of premature keratinization (95). Typically, the site at which the parakeratotic column arises lacks a granular layer, but elsewhere the granular layer is preserved. The histologic changes in the other forms of porokeratosis are similar but less pronounced than those seen in classic porokeratosis of Mibelli (Fig. 6-12).
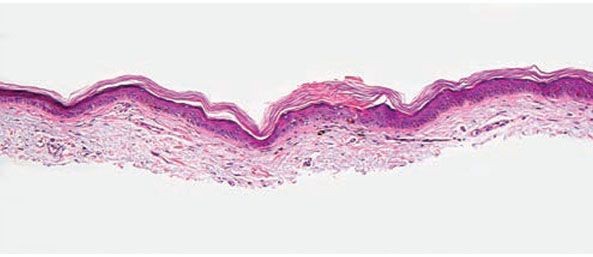
Figure 6-13 Porokeratosis. The cornoid lamella is a parakeratotic column overlying an effaced epidermis (original magnification ×100).
Because the peripheral raised ridge in porokeratosis slowly moves centrifugally, it stands to reason that the invagination is not bound to a definite structure, such as the sweat pore, as originally assumed by Mibelli. Although the invagination may be seen occasionally within a sweat pore or a pilosebaceous follicle, it is found most commonly in the epidermis independent of these cutaneous appendages (96). The epidermis overlying the central portion of a lesion of porokeratosis may be atrophic, normal in thickness, or rarely, acanthotic. A nonspecific perivascular infiltrate of chronic inflammatory cells is present in the dermis.
Transmission electron microscopic examination reveals signs of degeneration in keratinocytes beneath the parakeratotic column, with pyknotic nuclei, large perinuclear vacuoles that are separated from one another by cytoplasmic strands, and condensation of tonofilaments at their periphery (97). At the base of the parakeratotic column, dyskeratotic cells composed of nuclear remnants and aggregated tonofilaments are seen. The parakeratotic column is composed chiefly of cells with a pyknotic nucleus and a cytoplasm possessing high electron density because of the presence of many partially degraded organelles (98).
Differential Diagnosis. While the presence of cornoid lamella is essential for the diagnosis of porokeratosis, it is not a specific finding, and can be seen in other conditions including verruca vulgaris and actinic keratosis (99). The histologic differential diagnosis of punctate porokeratosis may include plantar or palmar verrucae. Clinical information, including age of onset, inheritance, and number and size of lesions, may be helpful in establishing a specific diagnosis. For discussion of porokeratotic eccrine duct nevus, see the section on nevus comedonicus.
Principles of Management. A variety of destructive therapies can be tried for cosmesis, including topical retinoids, topical 5-fluorouracil, cryotherapy, photodynamic therapy, or electrodessication. All patients with persistent porokeratosis should be monitored for the development of secondary skin cancers, particularly as they reach adulthood.
CONGENITAL BULLOUS DISORDERS
Epidermolysis Bullosa
Clinical Summary. Epidermolysis bullosa (EB) refers to a group of heritable skin fragility disorders caused by mutations in at least 18 genes (100). This group of disorders is further divided by the clinical, histologic, and electron microscopic findings. Four subcategories of EB are recognized: epidermal, junctional, dermal, and Kindler syndrome (Table 6-2) (101). In general, patients with EB develop blistering at site of minor friction or trauma. Identification of the subtype of EB is important to guide medical management for patients and provides valuable information for prognosis, surveillance, and genetic counseling in affected families.
Epidermolysis bullosa acquisita (EBA) is an immunobullous disorder that may present in childhood but more commonly in adults, and is fully discussed in Chapter 9.
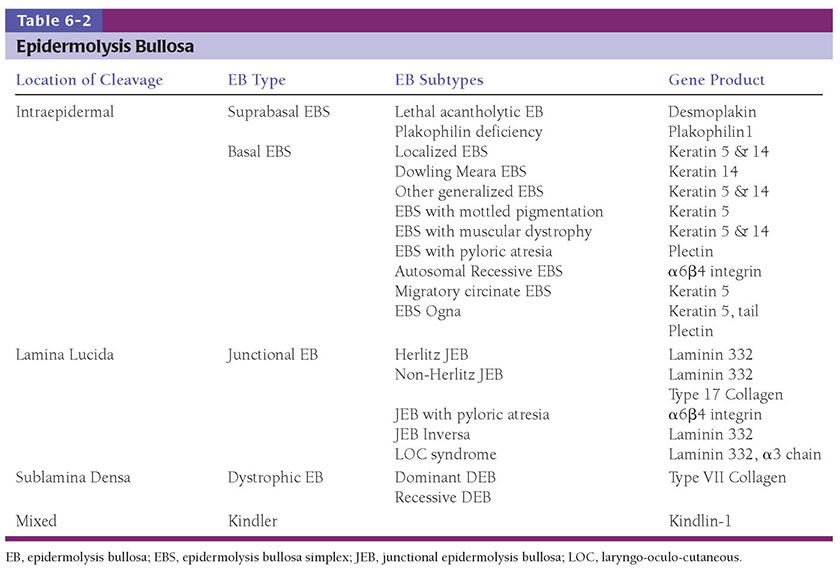
Epidermolysis bullosa simplex (EBS) refers to the EB subtypes demonstrating intra-epidermal bulla formation. Most patients present at birth but a small subset may not develop clinically appreciable skin fragility until adolescence. The cleavage plane is within the epidermis and healing occurs spontaneously without scarring (Fig. 6-14). EB simplex is further classified by extent of involvement and other associations (102). The Dowling-Meara variant may show generalized blistering, including involvement of the mucous membranes that at times is associated with mortality during early infancy (103,104) (see Fig. 6-23). High mortality is also seen in lethal acantholytic EB (LAEB). There is generalized epidermolysis of the skin, respiratory and gastrointestinal tracts (105).
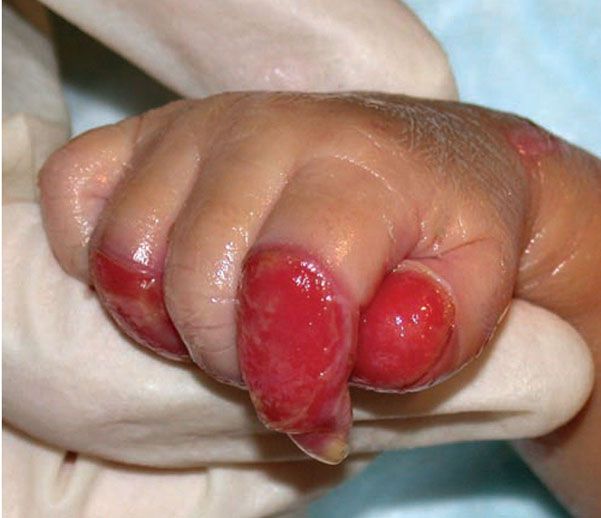
Figure 6-14 Epidermal bullosa simplex. Superficial blistering on the hand in a child with Epidermolysis bullosa simplex confirmed with genetic testing. (Image courtesy of CHOP dermatology.)
Junctional EB (JEB) presents with cleavage through the lamina lucida and therefore heals with scarring (Fig. 6-15). Widespread ulceration is often seen in the most severe variant Herlitz-JEB, and respiratory and gastrointestinal tract involvement leads to a high mortality (104). Less severe presentations have a higher survival rate. Periorificial blistering is particularly prominent in JEB with excessive granulation tissue noted in variants associated with laminin 332 mutation (106). Perioral granulation tissue associated with respiratory granulation tissue formation is characteristic of laryngo–onycho–cutaneous syndrome, also due to mutations in laminin 332. Patients with JEB have an especially high risk of highly aggressive squamous cell carcinomas (107,108).
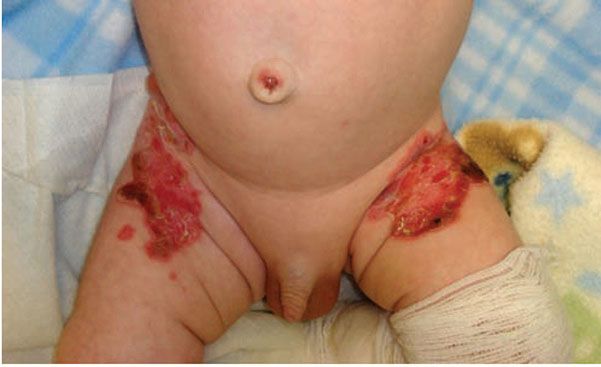
Figure 6-15 Junctional epidermolysis bullosa. Erosions and bullae in the diaper region, which characteristically heal with scarring. (Image courtesy of CHOP dermatology.)
Subepidermal blistering and ulceration is seen in dystrophic EB, including recessive and dominant variants. Dominant dystrophic EB has an acral predilection and often heals with milia and scarring. Recessive dystrophic EB is the most severe of the congenital bullous disorders. Blistering is generalized and patients may have severe involvement of the mucosal surfaces as well. Severe scarring leads to fusion of the fingers in toes, resulting in pseudosyndactyly (Fig. 6-16). Patients are at risk of recurrent skin infections and nutritional deficiency, which may necessitate placement of a feeding tube. In early adulthood, the ulcers and scars of the skin, mouth, and esophagus may give rise to aggressive squamous cell carcinomas (107). Rarely, EB may be transient and heal within a few months and has been termed bullous dermolysis in the newborn (109). It is likely that the majority of cases published as Bart’s syndrome, which was originally described as congenital absence of the skin (110), belong in this group.
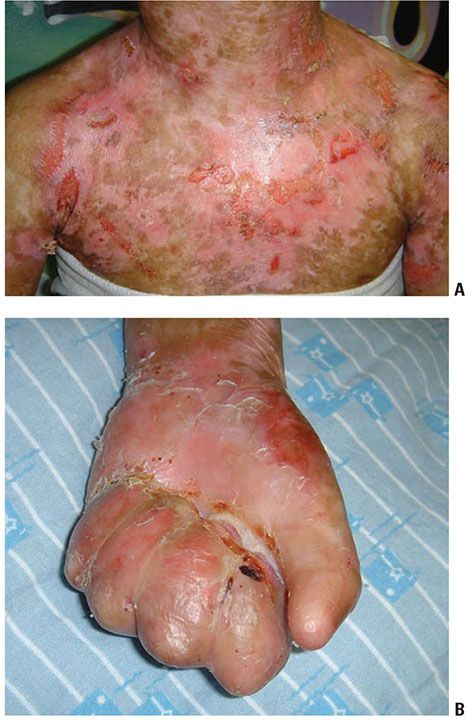
Figure 6-16 Recessive dystrophic epidermolysis bullosa. Child with generalized blisters and scarring (A) as well as mitten deformity of hands (B). (Image courtesy of CHOP dermatology.)
Similar to other forms of EB, neonates with Kindler Syndrome present with trauma-induced blistering most predominately in acral locations (111). The blistering dramatically improves or resolves with age. Photosensitivity with the development of poikiloderma is progressive. These cutaneous features are associated with mucosal involvement of the gingiva and anogenital mucosa. Severe colitis may result (112). Pseudosyndactyly and squamous cell carcinoma can be seen in areas of scarring but is less severe than in recessive dystrophic EB (113,114).
Histopathology. A biopsy is best taken from an induced blister because it will show a blister free of secondary changes. In an existing blister, the location of the blister may have changed as the result of regeneration of keratinocytes at the base of the blister or degeneration of the keratinocytes over the blister. The mode of artificially producing a blister depends on the degree of vulnerability of the skin. However, in most instances, gentle friction with a pencil eraser is used, or in older children, an activity that is known to induce blisters (115). Transmission electron microscopic examination has traditionally been the standard criterion for diagnosis of EB subtypes, but increased availability of immunofluorescence mapping techniques has added to the histologic diagnosis of EB and is able to guide more targeted genetic testing (116). Light microscopic features seen in the various forms of EB can be of additional diagnostic value. If possible, specimens of artificially induced blisters should be subjected to electron microscopic examination or immunofluorescence mapping to help guide diagnosis and therapy (115,117).
In EB simplex, the primary separation in induced blisters occurs within the basal cell layer. Spontaneously arising blisters may be found subepidermally as the result of complete disintegration of the basal cell layer. In bullae of more than a day’s duration, the cleavage may be found intraepidermally or subcorneally as a result of epidermal regeneration, thereby confusing the interpretation.
In JEB, the trauma of having a specimen taken for biopsy generally is sufficient to induce separation. This separation is located between the epidermis and the dermis (Fig. 6-17). In Herlitz–JEB, autopsy experience has revealed extensive subepithelial separation also in the gastrointestinal, respiratory, and urinary tracts. There are no morphologic or enzymatic abnormalities that can help distinguish the atrophic benign form of JEB from Herlitz–JEB.
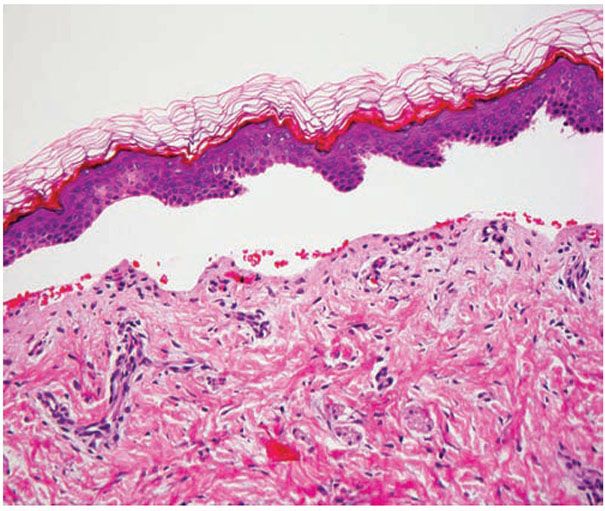
Figure 6-17 Junctional Epidermolysis Bullosa. A pauci-cellular subepidermal blister is present (original magnification ×200.)
In dominant dystrophic EB and recessive dystrophic EB, light microscopy shows dermal–epidermal separation. Scar tissue is a frequent finding in these biopsies.
The histology of Kindler syndrome depends on the characteristics of the lesion that is biopsied. A skin biopsy from an area of poikiloderma will show epidermal atrophy and loss of the rete ridges. Hyperkeratosis is variable (111,118). Pigmentary incontinence, blood vessel dilation, and elastosis may be found in the dermis. Abnormal distribution of melanosomes may be seen (118). A biopsy of a bullous area shows the cleavage plane on different levels.
The immunomapping procedure for diagnosis of EB consists of exposing cryostat sections to EB-relevant antibodies (101,119). The panel of antibodies used can define a more precise level of cleavage. Additional information is gained by looking at relative staining intensity of the targeted antibodies that can reveal the underlying molecular defects causing skin fragility in patients with EB (Fig. 6-18). Using an expanded panel of antibodies, higher sensitivity and specificity was demonstrated with immunofluorescence mapping over electron microscopy, in at least one center’s experience (119).
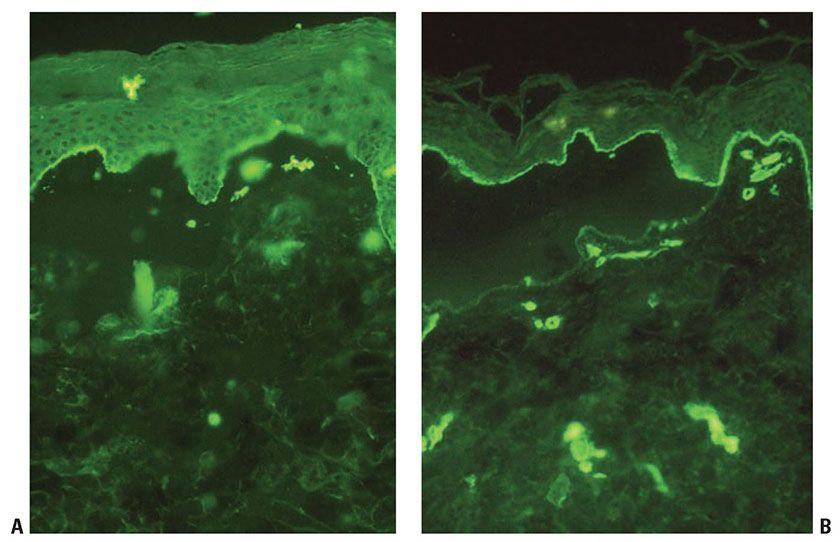
Figure 6-18 Immunofluorescence mapping for Epidermolysis Bullosa. In this case of dystrophic epidermolysis bullosa, BP180 (A) and laminin (B) can be seen on the roof of the blister (original magnification ×200). (These images are courtesy of Lori Prok, MD, University of Colorado and Children’s Hospital Colorado.)
Pathogenesis. In the epidermal types of EB, microscopic examination most frequently shows cleavage that is the result of degenerative cytolytic changes occurring in the lower portion of the basal cells between the dermal–epidermal junction and the nucleus. Immunofluorescence mapping shows that all three antigens (type IV collagen, laminin, bullous pemphigoid antigen) are located beneath the cleavage. Mutations of keratin genes for KRT5 and KRT14 on chromosome 12 and 17, respectively, have been demonstrated in EBS and these proteins can be demonstrated to be reduced or absent. The perturbation of keratin filament assembly results in clumping of tonofilaments in basal keratinocytes. Rarer subtypes of EBS (Table 6-2) have identified mutations in α6β4 integrin and plectin, resulting in poor integration of keratin filaments into hemidesmosomes (120). Intraepidermal cleavage is seen in subtypes caused by plakophillin and desmoplakin deficiency. EM examination demonstrates perinuclear retraction of keratin filaments (119).
In the junctional types of EB, electron microscopic examination often shows abnormal hemidesmosomes, especially in Herlitz–JEB. They may be reduced in size or number and may lack their subbasal cell-dense plaque (120). Immunofluorescence mapping shows type IV collagen and laminin on the floor of the blister. Bullous pemphigoid antigen is present mainly on the blister roof but also in a more spotty distribution and, to a much lesser extent, on the blister floor. Mutations have been found in any of three polypeptides of laminin 332 (formerly laminin 5): α-3 (LAMA3), BETA-3 (LAMB3), and GAMMA-2 (LAMC2). This mutation reduces adhesion between the epidermis and dermis (121). In Herlitz–JEB, laminin 332 is absent or more severely affected, leading to the more severe clinical phenotype. Non-Herlitz JEB may be caused by missense laminin 332, collagen 17, or α6β4 integrin mutations. Here, the relevant gene products are reduced on immunofluorescence (119). Electron microscopy shows more variability in the hemidesmosomal size and number (120).
On electron microscopy, the dermal types of EB show abnormalities in anchoring fibrils. Generalized recessive dystrophic EB demonstrates absence of the anchoring fibrils even in nonlesional skin, which is established through the lack of a reaction with monoclonal antibodies to the anchoring fibrils. Mutations in the gene encoding type VII collagen (COL 7A1) located at chromosome band 3p21 have been reported to cause these abnormal findings in the dystrophic forms of EB (122,123). Because type VII collagen is a major structural component of the anchoring fibrils, immunofluorescence staining with polyclonal antibodies to type VII collagen reveals complete absence of staining, even in the unaffected skin of patients with severe dystrophic recessive EB (119). Similarly, there is no reaction with periodic acid-thiosemicarbazide-silver proteinate, which stains anchoring fibrils selectively (124). In both dominant dystrophic EB and in localized recessive dystrophic EB, structurally normal anchoring fibrils are present but are reduced in number and function (125). Immunofluorescence mapping shows all three basement membrane zone constituents—bullous pemphigoid antigen, laminin, and type IV collagen—on top of the cleavage (119).
Kindler syndrome is unique among EB subtypes in that the layer of separation is variable. The FERMT1 gene, which is mutated, has important roles in keratinocyte migration, adhesion, and proliferation. This effect is at least in part through the activation of integrin function. Loss of FERMT1 activity leads to loss of cell adhesion as seen by electron microscopy as clefting within the basal keratinocytes, lamina lucida, or below the lamina densa. Hemidesmosomes and anchoring fibrils remain intact. There is disorganization of the basement membrane zone with characteristic reduplication of the of the lamina densa (126). Diminished integrin function may also account for epidermal atrophy through decreased epidermal proliferation.
Differential Diagnosis. Clinically, the distinction among the subtypes of EB can sometimes be challenging at birth. Diagnostic biopsies and genetic testing can aid in securing the diagnosis that is critical for management and prognostic significance. When presenting in a neonate, infectious etiologies of bulla must be considered excluded, including, but not limited to, congenital varicella, herpes simplex viral infection, and Staphylococcus aureus infection, leading to bullous impetigo or staph scalded skin syndrome. Autoimmune bullous disorders should also be considered. Infants born to mothers with pemphigus foliaceus, pemphigus vulgaris, and bullous pemphigoid may have transplacental passage of antigens and transient disease. Infants with epidermolytic hyperkeratosis may present with flaccid bullae or erosions that may mimic EB. Histologically, when confronted with a paucicellular vesicular process, one could consider entities such as cell poor bullous pemphigoid, porphyria, EBA, and friction blisters. However, clinical correlation and additional targeted testing as described above will assist in establishing a final diagnosis.
Principles of Management. The mainstay of EB management continues to be meticulous wound care and infection control. Nutritional support is important for the more severe forms. For scarring forms of EB, monitoring the development of cutaneous malignancy and conservative management when they do arise is of utmost importance. Allogeneic bone marrow transplant has been reported to improve fragility in some patients with recessive dystrophic EB (107). Experimental methods of gene correction and protein replacement for severe, scarring EB are emerging (127).
Familial Benign Pemphigus (Hailey–Hailey Disease)
Clinical Summary. Familial benign pemphigus is an autosomal dominant genodermatosis, with a family history obtainable in about two-thirds of patients. It presents in teenagers and young adults, usually at or after puberty, although presentation in infants has been infrequently described (128). It is characterized by a localized, recurrent eruption of small vesicles, erosions, and crusted plaques in the intertriginous areas. By peripheral extension, the lesions may assume a circinate configuration. The predilection of Hailey–Hailey for intertriginous areas, especially the axillae and the groin, likely has to do with the contribution of heat, moisture, and friction to the development of the erosions. Bacterial and candida superinfection is common. Rare instances of mucosal involvement have been reported, including the mouth, the labia majora, and the esophagus.
Histopathology. While early lesions may show small suprabasal separations, so-called lacunae, fully developed lesions show large separations in a predominantly suprabasal position (Fig. 6-19). Villi, which are elongated papillae lined by a single layer of basal cells, protrude upward into the bulla, and in some cases, narrow strands of epidermal cells proliferate downward into the dermis. Many cells of the detached stratum malpighii show loss of their intercellular bridges with acantholysis affecting large portions of the epidermis. Despite the extensive loss of intercellular bridges, the cells of the detached epidermis in many places show only slight separation from one another because a few intact intercellular bridges still hold them loosely together, leading to the so-called “dilapidated brick wall” appearance of the disease. Large numbers of individual and groups of cells are often seen in the bulla cavity.
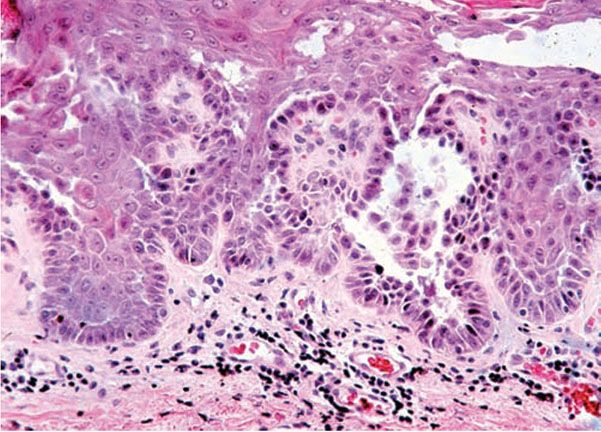
Figure 6-19 Familial benign pemphigus (Hailey–Hailey). The bulla is largely in a suprabasal position. The extensive loss of intercellular bridges with partial coherence of cells gives the detached epidermis the appearance of a dilapidated brick wall. On the right side within the granular layer, a corps rond can be seen (original magnification × 40).
Many of the cells of the stratum malpighii that have lost their intercellular bridges show a relatively normal cytoplasm and a normal nucleus in which mitotic activity is preserved. Some of the acantholytic cells, however, have a homogenized cytoplasm, suggesting premature partial keratinization. Such acantholytic cells with premature keratinization resemble the grains of Darier disease. Occasionally, a few corps ronds are present in the granular layer.
Pathogenesis. Genetic studies have localized the key mutations to the ATP2C1 gene on chromosome 3q, specifically 3q21-q24. Clinical variability is attributed to haploinsufficiency. This locus encodes the SPCA pumps that are responsible for ATP-dependent calcium transport along with the SERCA proteins that are mutated in Darier disease (129,130). The defective pump protein leads to decreased calcium content in the usually calcium-rich basal keratinocytes (131). It is hypothesized that the calcium defect disrupts protein processing leading to failure to process desmosomal proteins and therefore loss of cell-cell adhesion (129). Keratin expression and keratinocyte differentiation may also be affected (131). Exacerbation by physical stress such as heat and friction may lead to an additive decrease in SPCA expression.
Differential Diagnosis. Histologically, familial benign pemphigus shares certain features with both Darier disease and pemphigus vulgaris. In all three diseases, one finds predominantly suprabasal separation of the epidermis caused by acantholysis and resulting in lacunae or bullae and villi formation.
Several features can distinguish familial benign pemphigus from Darier disease because in Darier disease, the suprabasal separations usually are smaller, thus appearing as lacunae rather than as bullae; acantholysis is less pronounced, being limited to the lower epidermis, especially the suprabasal region; and dyskeratosis consisting of the formation of corps ronds and grains is much more evident.
Pemphigus vulgaris often resembles familial benign pemphigus, and the histologic differentiation of these two diseases may be impossible. There is generally less extensive acantholysis in pemphigus vulgaris, and it is limited largely to the suprabasal region. Therefore, the detached epidermis appears normal and lacks the appearance of a dilapidated brick wall. There is more severe degeneration of the acantholytic cells within and near the bulla cavity. The presence of eosinophils in the bulla points toward a diagnosis of pemphigus vulgaris, but their absence does not rule it out. Correlation of the findings on the routinely prepared sections with immunofluorescence and/or ELISA testing can establish a specific diagnosis.
Principles of Management. Reduction of triggering factors such as heat, friction, and moisture is the mainstay of treatment. Topical steroids and other anti-inflammatory preparations can be useful to reduce symptoms. Bacterial and fungal infections may exacerbate symptoms and require topical or systemic antimicrobial therapy.
Keratosis Follicularis (Darier Disease)
Clinical Summary. Darier disease is an autosomal dominant genodermatosis characterized by progressive hyperkeratotic or crusted papules in a seborrheic distribution (Fig. 6-20A, B). Peak age of onset is near the time of puberty, although congenital cases have been described (132). Crusted and coalescent plaques may form verrucous lesions. The oral mucosa may be involved (133). In some cases of Darier disease, keratotic papules that resemble those seen in acrokeratosis verruciformis of Hopf may be found on the dorsal hands and feet. Nails show longitudinal erythronychia, longitudinal leukonychia, and distal v-shaped nicks in the nail plate secondary to nail fragility. These nail findings are sometimes referred to as “candy cane nails.”
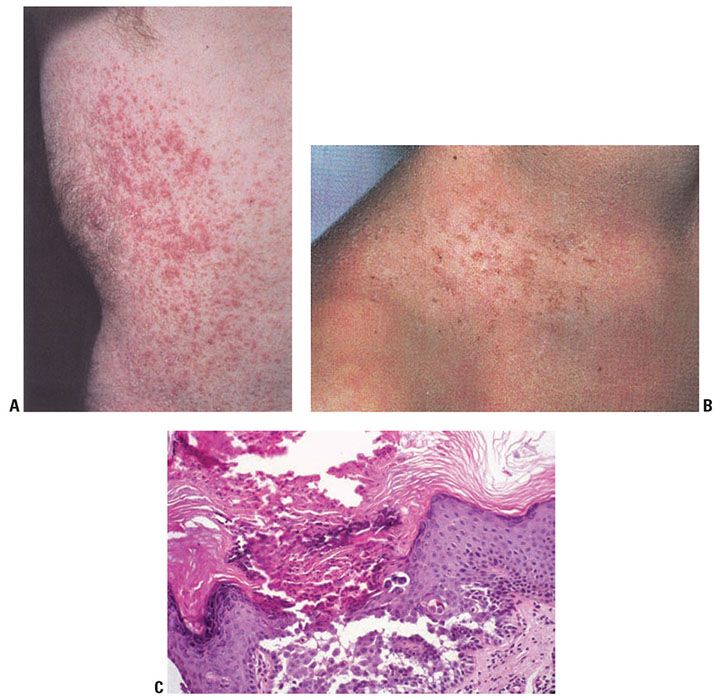
Figure 6-20 A: Darier disease. Multiple tiny hyperkeratotic papules that coalesce into plaques present on the trunk. B: Crusted papules in a follicular distribution at the base of the neck and upper chest. C: Darier disease, low magnification. Hyperkeratosis and papillomatosis are evident. Numerous lacunae are present. On the left are elongated papillae lined by a single layer of cells, so-called villi. Corps ronds are present in the granular layer, and grains are seen in the horny layer. The lacunae contain desquamated cells (original magnification ×100).
Keratosis follicularis may occur as a number of clinical variants including hypertrophic, vesiculobullous, and segmental Darier disease. In the hypertrophic type, widespread, markedly thickened, and hyperkeratotic lesions are seen, especially in the intertriginous areas. In the vesiculobullous type, vesicles and small bullae are seen in addition to papules. Segmental Darier disease is usually limited to one side in a blaschkolinear distribution and may be present at birth or acquired. The designation acantholytic dyskeratotic epidermal nevus has been suggested; however, mutations in ATP2A2 found in these specimens suggest that this is a form of mosaic Darier disease (134).
Histopathology. The characteristic changes in Darier disease include the following: a specific pattern of dyskeratosis resulting in the formation of corps ronds and grains; suprabasal acantholysis resulting in suprabasal clefts or lacunae; and irregular upward proliferation into the lacunae of papillae lined with a single layer of basal cells, so-called villi (Fig. 6-20C). There are also papillomatosis, acanthosis, and hyperkeratosis. The dermis shows a chronic inflammatory infiltrate. In some cases, there is downward proliferation of epidermal cells into the dermis.
The corps ronds occur in the upper stratum malpighii, particularly in the granular and horny layers. Grains are found in the horny layer and as acantholytic cells within the lacunae. Corps ronds possess a central homogeneous, basophilic, pyknotic nucleus that is surrounded by a clear halo. By virtue of size and the conspicuous halo, corps ronds stand out clearly (Fig. 6-21). Peripheral to the halo lies basophilic dyskeratotic material as a shell. The nonstaining halo in some instances is partially replaced by homogeneous, eosinophilic dyskeratotic material. Compared with the corps ronds, the grains are much less conspicuous. They resemble parakeratotic cells but are somewhat larger. The nuclei of grains are elongated and are surrounded by homogeneous dyskeratotic material that usually stains basophilic but may stain eosinophilic. The lacunae represent small, slit-like intraepidermal vesicles most commonly located directly above the basal layer. They contain acantholytic cells and show premature partial keratinization. Because of shrinkage, some of them are elongated, and these then appear identical with the grains in the horny layer. The villi projecting into the lacunae may be quite tortuous, so on histologic examination, some of them appear in cross section as rounded dermal structures lined by a solitary row of basal cells.
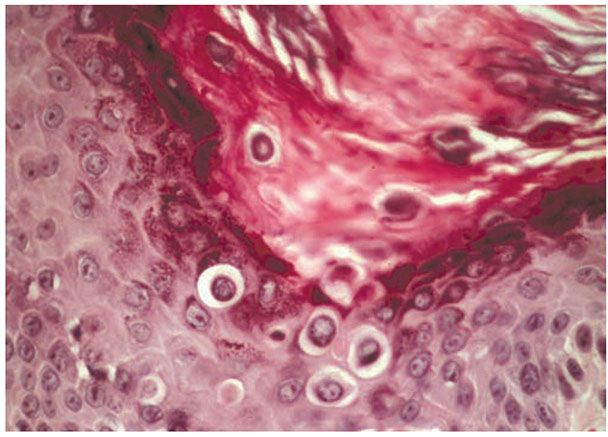
Figure 6-21 Darier’s disease. Hyperkeratosis is present with corps ronds in the thickened stratum corneum and epidermis. Suprabasalar acantholysis is present (original magnification ×400).
Hyperkeratosis and papillomatosis may cause the formation of keratotic plugs, which often fill the pilosebaceous follicles but are also found outside of follicles. Darier disease is not exclusively a follicular disorder, which is highlighted by the fact that areas devoid of follicles, such as palms, soles, and the oral mucosa, may be affected.
In hypertrophic lesions of Darier disease, considerable acanthosis may occasionally be observed, either as proliferations of basal cells or as pseudocarcinomatous hyperplasia. Proliferations of basal cells consist of long, narrow cords composed of two rows of basal cells separated by a narrow lacunar space.
The vesiculobullous lesions, which occur in rare instances, differ from lacunae merely in size; they contain numerous shrunken cells with the appearance of grains.
The keratotic papules that may occur on the dorsa of the hands and feet, which clinically resemble those seen in acrokeratosis verruciformis of Hopf, show mild dyskeratotic changes and often suprabasal clefts on serial sectioning. They are a manifestation of Darier disease and not of acrokeratosis verruciformis.
The lesions on the oral mucosa are analogous in appearance to those observed on the skin and thus show lacunae and dyskeratosis, although definite well-formed corps ronds generally are absent.
Pathogenesis. Darier disease is due to mutations in the ATP2A2 gene, which encodes the sarcoplasmic/endoplasmic reticulum calcium pumping ATPase (SERCA2) on chromosome 12q23-24.1. Diminished functioning of this ubiquitous protein leads to decreased calcium content in the stratum basalis (131). Perturbation of the intracellular calcium disrupts calcium-dependent signaling, resulting in loss of adhesion and dyskeratosis. In addition, transduction of SERCA2 mutant protein into human keratinocytes caused cells to detach and be resistant to apoptosis in the face of a second endoplasmic reticulum stress, explaining a possible gain-of-function mechanism for the acantholysis and dyskeratosis in Darier disease (135).
Acantholysis has been thought by some authors to be due to the loss of the intercellular contact layer within desmosomes, both in Darier disease and in familial benign pemphigus. The two halves of the desmosomes then pull apart, after which the tonofilaments become detached from them. Another group of authors believes that there is some basic defect in the tonofilament–desmosome complex in Darier disease and in familial benign pemphigus, resulting in separating of tonofilaments from the attachment plaques of desmosomes. It is likely that both processes take place simultaneously in both Darier disease and familial benign pemphigus.
The cause for the acantholysis in Darier disease and familial benign pemphigus is not definitely known yet. Faulty synthesis of the intercellular substance has long been suspected. Further studies have suggested that intercellular communication is crucial for epidermal differentiation. This was corroborated when mutations in ATP2A2 were found to cause Darier disease and disclosed a role for the SERCA2 pump in the calcium signaling pathway that regulates cell-to-cell adhesion and differentiation of the epidermis (136).
Loss of calcium homostasis via inhibition of SERCA2 is sufficient to disrupt desmosome assembly and lead to loss of intracellular adhesion (137). In association with the loss of desmosomes, excessive amounts of tonofilaments form within the keratinocytes around the nucleus as thick, electron-dense bundles. A defect of the tonofilaments would best explain the dyskeratotic features of both Darier disease and familial benign pemphigus. In Darier disease, the dyskeratosis is much more pronounced than in familial benign pemphigus, and thick bundles of tonofilaments, often in association with large keratohyaline granules, form large aggregates of homogenized dyskeratotic material. The corps ronds, on electron microscopic examination, are characterized by extensive cytoplasmic vacuolization. They show, in their center, an irregularly-shaped nucleus surrounded by a halo of autolyzed electron-lucid cytoplasm and, at their periphery, a shell of tonofilaments. On electron microscopic examination, the grains are seen to consist of nuclear remnants surrounded by dyskeratotic bundles of tonofilaments.
Whereas histologically a distinction between Darier disease and familial benign pemphigus is generally possible, with dyskeratosis being the predominating factor in Darier disease and acantholysis in familial benign pemphigus, this distinction is not as clearly evident in electron microscopic examination. The reason for this is that for electron microscopy, only a small specimen can be processed, which in either of the two diseases shows predominantly acantholysis in some instances and dyskeratosis in others and only rarely shows both. In both diseases, however, acantholysis precedes dyskeratosis.
In familial benign pemphigus, too, after loss of the desmosomes, excessive amounts of tonofilaments form within the keratinocytes and aggregate around the nucleus as thick, electron-dense bundles, often in a whorling configuration. However, even though dyskeratosis is present, it is less pronounced than in Darier disease, and most of the keratinocytes keratinize normally, with only very few becoming grains or corps ronds as the result of dyskeratotic degeneration.
Differential Diagnosis. Although acantholytic dyskeratosis in association with corps ronds is highly characteristic of Darier disease, it also occurs in several other conditions: in warty dyskeratoma, a solitary lesion with a deep central invagination; in transient or persistent acantholytic dermatosis (Grover disease), in which the lesions consist of discrete papules; in focal acantholytic dyskeratoma, manifesting itself as a solitary papule; and as an incidental small focus in a variety of unrelated lesions. Occasionally, a few corps ronds are also seen in familial benign pemphigus.
Principles of Management. Topical or systemic retinoids can effectively control Darier disease. Topical corticosteroids can also be helpful. Bacterial or fungal overgrowth may be treated with topical antimicrobial preparations.
MISCELLANEOUS CONDITIONS
Acrokeratosis Verruciformis of Hopf
Clinical Summary. In acrokeratosis verruciformis (AKV), an autosomal dominant disorder, numerous flat, hyperkeratotic, occasionally verrucous papules are present on the distal part of the extremities, predominantly on the dorsa of the hands and feet (Fig. 6-22
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


