Fig. 6.1
Schematic representation of the hemidesmosomal adhesion complex. (a) A transmission electron micrograph (kindly provided by Dr. Ingrid Hausser) is shown; the main molecular components of the hemidesmosomal adhesion complex are superimposed as a drawing. (b) Schematic representation of the full-length and shed forms of collagen XVII. The interacting proteins are in bold, and the proteases involved in the cleavage of the ectodomain are in italics. Collagenous domains are in pink. IC intracellular, EC extracellular, NC16A non-collagenous domain 16A
Collagen XVII is expressed in the epidermis of the skin and in the epithelia of oral mucosa, ocular conjunctiva and cornea, upper oesophagus and transitional epithelium of the bladder [1, 6, 7]. Some investigations have found low-level expression also in certain cell populations in the kidney, brain, placenta and amniotic membranes, as well as in the developing heart and teeth [1, 6, 7]. Recently, collagen XVII was reported to be highly expressed in hair follicle stem cells [8]. Analysis of Col17a1-deficient mice, which show premature hair greying and hair loss, suggested that collagen XVII is critical for the self-renewal of both hair follicle stem cells and melanocyte stem cells [8].
The collagen XVII molecule is a type II transmembrane protein. It is a homotrimer consisting of three 180 kDa alpha-1 (XVII) chains, each with a globular N-terminal intracellular domain, a short hydrophobic transmembrane stretch and an extracellular C-terminus (Fig. 6.1b) [9]. The intracellular domain, part of the hemidesmosomal plaque, has no structural similarities to other proteins, and it interacts with the integrin β4 subunit, plectin and BP230. The extracellular domain contains 15 collagenous subdomains interrupted by short non-collagenous stretches. The juxtamembranous non-collagenous region NC16A is likely to be important for trimerisation and subsequent triple-helix folding in N- to C-terminal direction [10]. It also harbours the epitopes recognised by autoantibodies in bullous pemphigoid and pemphigoid gestationis (reviewed in [11]).
Collagen XVII contributes to the structure of anchoring filaments in the lamina lucida of the epidermal basement membrane and contains at least one loop structure in the lamina densa. Its binding ligands are laminin 332 and the integrin α6 subunit [1, 2, 12] (Fig. 6.1a, b). The extracellular domain can undergo proteolytic shedding resulting in the formation of a 120 kDa fragment and subsequent cleavage to a shorter soluble form of 97 kDa (Fig. 6.1b). The constitutive shedding, which results in the 120 kDa ectodomain, is mediated by ADAM-9, ADAM-10 and ADAM-17 [13–15]. This shedding seems to be dependent on the conformation of the NC16A domain and the steric availability of the cleavage site [16]. Its regulation is complex: the plasma membrane microenvironment, i.e. the organisation of lipid rafts [17], and the extracellular phosphorylation of collagen XVII by ecto-casein kinase 2 [18] can modulate shedding. The cleavage of collagen XVII occurs within the NC16A domain, and various N-termini have been identified between the amino acid residues 514 and 547 [19]. There is evidence that the shed ectodomain is incorporated in the basement membrane and may have cell adhesion properties [20]. The cleavage process itself presumably regulates keratinocyte detachment from the basement membrane in the process of cell differentiation and migration [13]. The C-terminal cleavage of the extracellular domain into a 97 kDa fragment seems to depend, at least in vitro, on plasmin [21]. Its biological relevance is not well understood. Nevertheless, shedding is clinically relevant because the 120 kDa ectodomain and the 97 kDa form are targets for autoantibodies in the autoimmune blistering diseases (reviewed in [11]).
6.2 Junctional EB
JEB is a clinically and genetically heterogeneous type of epidermolysis bullosa, characterised by mechanically induced tissue separation along the lamina lucida of the basement membrane. Clinical symptoms are skin and mucosal blistering, with or without involvement of other organs. The molecular basis of JEB is complex; mutations in seven different genes can cause this EB type. The genes encode proteins which are functionally related and interact closely with each other—laminin 332, collagen XVII, integrin α6β4 and the integrin α[alpha]3 subunit [22, 23]. JEB comprises two main phenotypes: an early lethal phenotype, designated as JEB generalized severe, also known as JEB Herlitz, which is caused by complete loss of laminin 332, and a spectrum of less severe phenotypes, collectively called as JEB-non Herlitz or JEB generalized intermediate or JEB-other, which are caused by mutations in the genes encoding laminin 332, collagen XVII or integrin α[alpha]6β[beta]4 [5].
6.3 The Clinical Spectrum of JEB Generalized Intermediate
The group of JEB generalized intermediate was historically split into several subtypes, based on clinical criteria: generalised, localised or late onset. These descriptive terms aim at pointing to the major features of the respective subtypes. However, they refer to different characteristics of the disease—distribution of the lesions, course of the disease—which makes their use difficult and obsolete. Furthermore, the clinical distinction between JEB generalized intermediate caused by mutations in the genes for laminin 332 and collagen XVII is subtle and often not possible. Therefore, a collective designation such as JEB generalized intermediate avoids unnecessary heterogeneity and is practical.
The full-blown clinical picture of JEB generalized intermediate caused by loss of collagen XVII (Fig. 6.2a, b) comprises congenital generalised blistering and other progressive signs which develop with advancing age, such as skin atrophy and dyspigmentation, dystrophy and loss of nails and alopecia. Mucosal involvement is mild and may be oral, ocular, nasal and genitourinary. Teeth are always affected by amelogenesis imperfecta, manifesting as enamel pits and by increased incidence of caries. Melanocytic nevi at site of prior blisters—epidermolysis bullosa nevi—have been first described in this subtype but later reported in all EB types [24]. Heterozygous carriers of collagen XVII mutations may have enamel defects, demonstrating that reduced amounts or mutated collagen XVII may be sufficient for dermal-epidermal stability, but not for proper dental development [25].
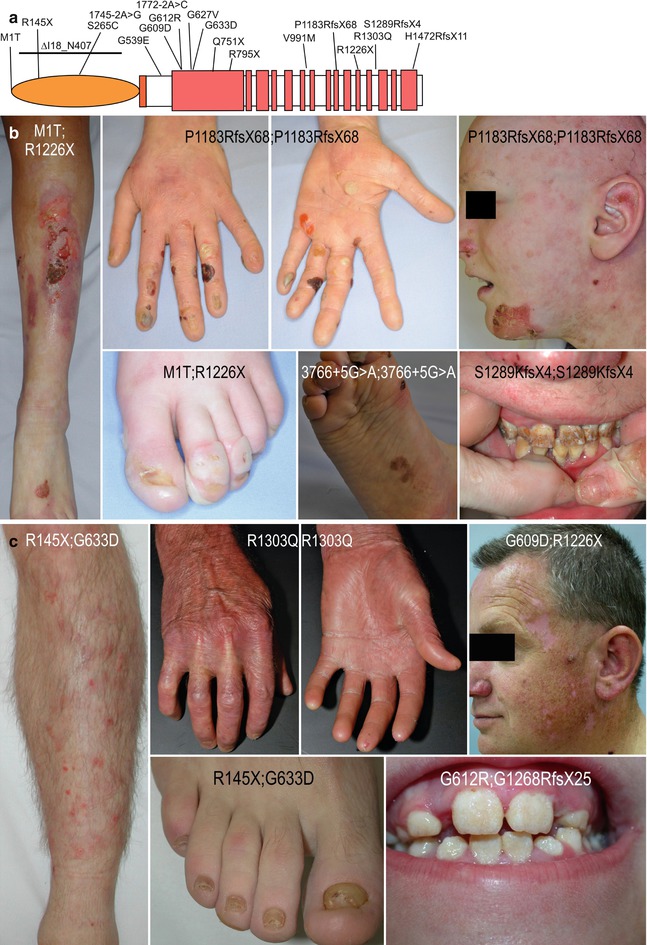
Fig. 6.2
Collagen XVII mutations and associated phenotypes. (a) Schematic representation of the collagen XVII molecule with the mutations discussed in this review. (b) Clinical features of JEB generalized intermediate due to COL17A1 null mutations and complete loss of collagen XVII: widespread, almost permanent blistering, wounds, haemorrhagic crusts, nail dystrophy or loss, epidermolysis bullosa nevus, alopecia and severe enamel hypoplasia. (c) Mild clinical features in patients with residual expression of mutant collagen XVII: minor blistering, skin atrophy, loss of dermatoglyphs, toenail dystrophy and enamel pitting, but no hair anomalies
Compared to the phenotype of collagen XVII null mutations, as described above, milder disease is associated with mutations leading to expression of residual amounts or mutant (either truncated or with amino acid substitutions) collagen XVII. In these cases, skin fragility is milder, with fewer blisters and, sometimes, later onset. Fingernails may be normal or dystrophic, but toenails are mostly dystrophic (Figs. 6.2a, c). Teeth are usually affected, but hair and mucous membranes are not. Various symptom combinations exist, but these are usually not predictable on the basis of the mutation constellation.
6.4 The Spectrum of Collagen XVII Mutations and Genotype-Phenotype Correlations
The alpha1-(XVII) chain is encoded by the COL17A1 gene which spans 52 kb of the genome and is located on the long arm of chromosome 10 (10q24.3). COL17A1 has a split structure consisting of 56 exons (most of them in frame) and short introns. Thus far, 80 different COL17A1 mutations have been reported in the literature. They include nonsense mutations, splice site mutations, deletions and insertions. Most of them lead to formation of premature termination codons, mRNA decay and absence of collagen XVII expression [25–28]. However, the consequences and phenotypes of the mutations are difficult to predict and require analyses on mRNA and protein level. In some cases, nonsense mutations can cause mild phenotypes because of alternative splicing mechanisms. For example, the mutation p.R795X in exon 33—a frequent mutation in Italian patients—led to alternatively spliced COL17A1 mRNA that entirely lacked exon 33. This allowed residual synthesis and expression of somewhat shorter collagen XVII at the cutaneous basement membrane zone [29]. Similarly, in a mildly affected patient with the mutation p.Q751X, the deleterious effect was skirted by deleting exon 30 containing the premature termination codon. The reading frame was restored, resulting in a shorter transcript that deleted 12 amino acids from the collagenous 15 (Col15) domain [30].
The consequences of splice site mutations are hardly predictable without RNA and protein studies. Some interesting constellations have been reported. Certain splice site mutations allow residual expression of truncated or full-length collagen XVII ([27, 31]). For example, the mutation c.1745–2A>G at the acceptor splice site of exon 21 was found in three siblings over 70 years of age with very mild skin fragility and toenail dystrophy as only symptoms. Truncated collagen XVII was found in the skin at a level corresponding to about 15 % of normal, but this was sufficient for relatively stable dermal-epidermal adhesion [27]. Surprisingly, c.1772–2A>C, at the acceptor splice site of exon 22, which codes for a segment of the extracellular domain of collagen XVII, has been reported to cause intraepidermal cleavage at the level above the cytoplasmic attachment plaque of the hemidesmosomes (Fig. 6.2a) [32]. A similar constellation was described in the case of the 1,172 base pairs in-frame deletion, p. ΔI18_N407, which deleted a large segment from the intracellular domain of collagen XVII (Fig. 6.2a) and led to intraepidermal skin cleavage and phenotypic features of epidermolysis bullosa simplex [33].
The deletion of c.4410_4413dupCATT, p.H1472RfsX11 (Fig. 6.2a) was shown to truncate the C-terminus and to impair N-glycosylation of the ectodomain. This led to intracellular accumulation, indicating that N-glycosylation of the ectodomain is required for targeting of collagen XVII to the plasma membrane [34].
Only nine missense mutations in the COL17A1 gene have been reported. There are no clear-cut genotype-phenotype correlations for these mutations, but patients seem to have a milder disease than patients with null mutations [35] (Fig. 6.2). The glycine substitutions in the Col15 domain, p.G609D, p.G612R, p.G627V and p.G633D, have been carefully investigated. As in the case of other collagens, glycine substitutions interrupt triple-helix formation and lead to partial unfolding of the ectodomain, which can cause intracellular accumulation, affect posttranslational modifications and render the ectodomain on the cell surface susceptible to tissue proteolysis [35–38]. Interestingly, it was suggested that the mutation p.G627V could act in a dominant fashion, giving rise to severe dental enamel hypoplasia or JEB generalized intermediate in heterozygous patients [39–41]. However, the authors did not exclude a heterozygous large deletion on the second allele, which cannot be detected by routine mutation detection strategies [41].
Very few amino acid substitutions have been found in the non-collagenous domains, among them p.S265C, p.G539E, p.V991M and p.R1303Q (Fig. 6.2a); their molecular consequences remain elusive. In a Chinese patient, the homozygous variant p.S265C probably prevented targeting of collagen XVII into the plasma membrane, since no fluorescent signal with an antibody to the extracellular domain was observed. Also, the clinical picture was reminiscent of null mutations, i.e. generalised blisters since birth and severe alopecia since childhood [42]. The missense mutation p.G539E allowed synthesis of immunoreactive collagen XVII in keratinocytes but prevented its secretion, thus causing lack of the protein in the skin and a severe phenotype [43]. The role of the variant p.V991M remains to be clarified, since it was assigned as a single-nucleotide polymorphism (rs138824013) [44]. The mutation p.R1303Q is associated with a particular phenotype. We and others observed several patients with the same characteristics: late onset of mild skin fragility, pronounced atrophy of the skin with absence of dermatoglyphs, sclerodermiform appearance of the hands (Fig. 6.2c), enamel pits and normal hair. Immunofluorescence mapping demonstrated preserved collagen XVII expression and irregular, thickened staining pattern of laminin 332 and collagen VII [45, 46].
6.5 Molecular Diagnostics of JEB Generalized Intermediate
Clinical diagnosis of JEB generalized intermediate is practically impossible in newborns due to absence of secondary signs [47]. The situation may be easier in adults with a fully developed phenotype. Molecular diagnosis is recommended in all cases in order to achieve precise diagnosis for prognostication and prenatal diagnosis. Dermatohistopathology of skin biopsies is not helpful due to insufficient resolution of light microscopy.
Immunofluorescence mapping is the method of choice for diagnostics of all EB forms. This method makes use of a panel of antibodies against components of the dermal-epidermal junction zone, and it reveals the level of tissue separation within the blistering areas. Often, absent or attenuated staining of a particular protein can suggest the candidate gene [48]. JEB generalized intermediate is characterised by junctional blistering with collagen IV and collagen VII signals at the blister base and cytokeratin and plectin signals at the blister roof. Loss of collagen XVII expression is a diagnostic sign for JEB generalized intermediate. However, secondary reduction of signals of interacting proteins may present a diagnostic challenge. In cases with mild skin fragility, multiple components may exhibit reduced staining, without clear indication of the primary genetic defect. In these cases, several genes have to be screened for mutations. Transmission electron microscopy was previously employed as a common diagnostic method. However, it is tedious and time consuming and not used very much anymore. Typical ultrastructural changes in JEB generalized intermediate skin include cleavage within the lamina lucida and hemidesmosomes, which appear either normal or decreased in number and size.
Finally, mutation analysis is the gold standard method, which discriminates between different genetic defects. In cases with clear-cut immunofluorescence mapping results and typical clinical presentation, the candidate gene approach can be successfully used. In cases with mild skin fragility, the staining for several interacting proteins may be attenuated, which makes mutation analysis of many genes laborious and sometimes frustrating. In future, exome sequencing is likely to become a common method for mutation screening for heterogeneous genetic skin diseases, including JEB generalized intermediate. The correct molecular diagnosis is particularly important for the newborn, since it can predict the prognosis with relatively high probability. Nevertheless, rare, unexpected constellations exist. By establishing the molecular consequences of the individual mutations, phenotype-genotype correlations can be elucidated. Once the mutations disclosed, prenatal and preimplantation genetic diagnostics are important options for families at risk for severe disease.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


