(1)
Department of Dermatology, Columbia University, New York, NY, USA
(2)
Department of Dermatology, Morgan Stanley Children’s Hospital of New York-Presbyterian/Columbia University, New York, NY, USA
34.1 Introduction
34.2 History
34.3 Clinical Features
34.4 Clinical Course
34.5 Diagnosis
34.6 Disease Pathogenesis
34.7 Prognosis
The authors have no funding support to disclose.
34.1 Introduction
Chronic bullous disease of childhood (CBDC), also known as linear IgA disease (LAD), is the most common acquired autoimmune blistering disorder in children. It shares the diagnostic linear IgA staining of the basement membrane zone (BMZ) on direct immunofluorescence with adult LAD; however, the clinical features of CBDC distinguish it from other entities.
34.2 History
In the historical literature, the terms “bullous dermatitis herpetiformis,” “juvenile dermatitis herpetiformis,” “bullous pemphigoid of childhood,” and “linear IgA dermatosis of childhood” have all been used to describe cases of what is currently known as CBDC [1]. CBDC was actually initially described as a variant of dermatitis herpetiformis (DH) in the late nineteenth century [2]. It was later recognized as clinically distinct from DH by Kim and Winkelmann in 1961 [3] and ultimately differentiated as a unique disorder in 1970 by Jordon et al. who proposed the term “benign chronic bullous dermatosis of childhood” [4]. It was not until 1988 that CBDC was recognized as the childhood counterpart of adult linear IgA disease [5, 6].
34.3 Clinical Features
CBDC usually occurs in children 6 months to 10 years of age with a mean age of 4.5 years [6]. There are several case reports of neonatal CBDC, some presenting as early as 24–48 h after birth [7–9]. Rarely, onset occurs after the first decade [10]. It seems to occur in equal frequencies in boys and girls and there is no known race predilection.
The onset of the eruption is abrupt with development of tense, clear, or hemorrhagic vesicles and bullae on normal or erythematous skin. The lesions tend to occur in an annular array. New lesions may arise around resolving lesions, with arciform bullae surrounding a central crust, forming a “rosette pattern.” These distinctive lesions have also been described as a “string of pearls” or “cluster of jewels.” Clinical symptoms of CBDC range from asymptomatic to severe pruritus. Some children report a burning sensation.
The location of the eruption often depends upon the age of the patient. In younger children, the location is classically facial and perineal. On the face, lesions tend to occur in a perioral pattern (Fig. 34.1). Of note, disease onset in the perineum has been mistaken for sexual abuse [11]. Older children are more likely to present with a generalized eruption [6] (Fig. 34.2). Mucous membrane involvement, including oral vesicles, ulcers, or a desquamating gingivitis as well as conjunctival disease resulting in irritation, vision abnormalities, and scarring, may occur [12, 13].
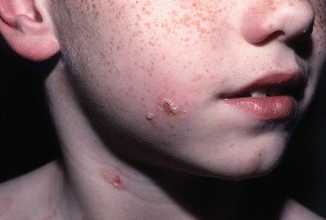
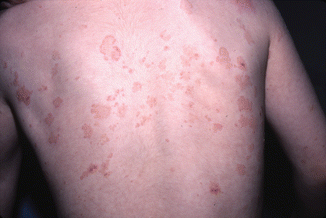
Fig. 34.1
Tense bulla arranged in a “string of pearls” distribution (Courtesy of Dr. Maria Garzon. Reprinted from Dermatology Clinics, Autoimmune Blistering Diseases: Part I – Pathogenesis and Clinical Features, 29/3, Mintz EM, Morel KD, Clinical Features, Diagnosis, and Pathogenesis of Chronic Bullous Disease of Childhood, Pages 459–462, (2011) with permission from Elsevier)
Fig. 34.2
Erythematous annular plaques and crusted erosions (Courtesy of Dr. Maria Garzon. Reprinted from Dermatology Clinics, Autoimmune Blistering Diseases: Part I – Pathogenesis and Clinical Features, 29/3, Mintz EM, Morel KD, Clinical Features, Diagnosis, and Pathogenesis of Chronic Bullous Disease of Childhood, Pages 459–462, (2011) with permission from Elsevier)
34.4 Clinical Course
CBDC is often initially misdiagnosed as bullous impetigo and may seem to improve with a brief course of systemic antibiotics. However, inevitably, the eruption will persist or recur, prompting further workup and concluding with the correct diagnosis. It is also not unexpected to find a positive wound culture of a lesion as they may become colonized or even superinfected with bacteria. The disease generally requires treatment as described in Chap. 57. Although the expected natural history is eventual remission, untreated, the disease is associated with significant morbidity. Neonatal CBDC has been associated with severe mucosal involvement and respiratory failure [7, 9]. Permanent sequelae, including blindness [7] and dysphagia [8], have been described. The goal of treatment is to control the disease symptoms while minimizing medication side effects.
Eventual remission occurs after several months to up to 4 years after onset. Rare cases have been described beyond 4 years although it usually resolves by puberty, with a few persisting into late adolescence or adulthood [6, 14]. Generally, repeat biopsy should be considered to review the diagnosis for persistent cases. Postinflammatory pigmentary alteration is common but usually not permanent. Scars are not expected in routine cases.
34.5 Diagnosis
In addition to the common initial clinical suspicion for bullous impetigo, the differential diagnosis of CBDC includes epidermolysis bullosa acquisita and other autoimmune bullous disorders such as bullous pemphigoid. In the neonatal period, it may be difficult to distinguish from genetically inherited blistering disorders, especially when the family history is noncontributory.
Histology as well as immunofluorescent testing is required to confirm the diagnosis. Histologic examination reveals a subepidermal blister with a predominantly neutrophilic infiltrate in the papillary dermis. Mononuclear cells and occasional eosinophils can also be seen (Fig. 34.3). Immunofluorescent testing of perilesional skin is the gold standard for diagnosis and is characterized by linear IgA staining of the BMZ (Fig. 34.4). For unclear reasons, it is recommended that one should avoid using the volar forearm as the site for biopsy, as immunofluorescent testing in this location may be falsely negative [15]. On salt split skin, autoantibodies bind to the epidermal side in most patients; however, antibody binding to the dermal side or to both the epidermal and dermal side occurs in some cases [16]. Circulating autoantibodies are of the IgA1 subclass [17] and can be found in over 90 % of patients [18]. A subset of patients manifest clinically as CBDC but show a mixed pattern of IgA and IgG antibodies on immunofluorescent testing; this entity has been termed “mixed immunobullous disease of childhood” and is similar to CBDC in terms of target antigens, disease presentation and course, and response to treatment [19].

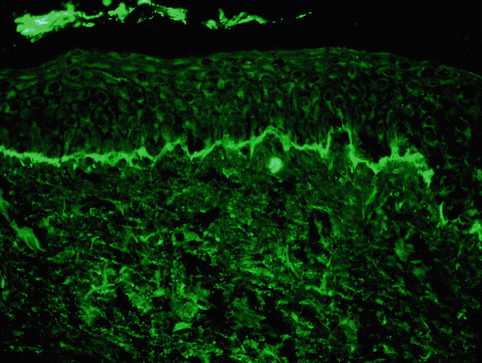
Fig. 34.3
Biopsy specimen showing a subepidermal split with a predominantly neutrophilic infiltrate in the papillary dermis (high power, hematoxylin and eosin stain) (Courtesy of Dr. Sameera Husain. Reprinted from Dermatology Clinics, Autoimmune Blistering Diseases: Part I – Pathogenesis and Clinical Features, 29/3, Mintz EM, Morel KD, Clinical Features, Diagnosis, and Pathogenesis of Chronic Bullous Disease of Childhood, Pages 459–462, (2011) with permission from Elsevier)
Fig. 34.4
Direct immunofluorescence demonstrating linear deposition of IgA along the basement membrane zone (Courtesy of Dr. John Zone, University of Utah. Reprinted from Dermatology Clinics, Autoimmune Blistering Diseases: Part I – Pathogenesis and Clinical Features, 29/3, Mintz EM, Morel KD, Clinical Features, Diagnosis, and Pathogenesis of Chronic Bullous Disease of Childhood, Pages 459–462, (2011) with permission from Elsevier)
34.6 Disease Pathogenesis
The exact etiology of CBDC is unclear. CBDC is associated with autoimmune haplotypes HLA-B8, Cw7, and DR3, suggesting there may be an underlying genetic susceptibility to this acquired disorder [20]. Most cases are idiopathic but CBDC may be triggered by exogenous factors such as infections, drugs, vaccinations, ultraviolet radiation, or malignancy [12, 21]. Such events may initiate an unknown sequence of changes that cause a normal component of the BMZ to become antigenic [5]. Autoantibodies are most often directed against proteolytic fragments of collagen XVII, a hemidesmosomal transmembrane protein also referred to as the 180 kd bullous pemphigoid antigen (BP180) [22]. The 120 kd shed ectodomain of collagen XVII has been recognized as a key autoantigen in adult linear IgA disease (and subsequently CBDC) and was termed “LAD-1,” the linear IgA disease antigen [23]. This 120 kd protein is further processed into a 97 kd protein, another principal autoantigen in CBDC [24, 25]. The full-length collagen XVII protein (BP180) is targeted by IgA in CBDC as well but less efficiently than the 120 kd and 97 kd protein fragments. Other autoantibodies are rarely identified including BP230 [26], collagen VII [27, 28], and antigens of molecular weights 145 kd [29], 200 kd [30], 255 kd [31], 285 kd [32], and 290 kd [33]. Immunoelectron microscopy has also demonstrated antibody labeling of heterogeneous components of the BMZ.
Related posts:
 Kindlin-1 and Its Role in Kindler Syndrome
Kindlin-1 and Its Role in Kindler Syndrome
 Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
 Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
 Living with Epidermolysis Bullosa: Reviewing the Impact on Individuals’ Quality of Life
Living with Epidermolysis Bullosa: Reviewing the Impact on Individuals’ Quality of Life
 How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


